酶(enzyme)是活细胞内产生的具有高度专一性和催化效率的蛋白质,又称为生物催化剂,生物体在新陈代谢过程中,几乎所有的化学反应都是在酶的催化下进行的。
细胞内合成的酶主要是在细胞内起催化作用,也有些酶合成后释入血液或消化道,并在那里发挥其催化作用,人工提取的酶在合适的条件下也可在试管中对其特殊底物起催化作用。
酶学知识来源于生产实践,我国4千多年前的夏禹时代就酿酒盛行,周朝已开始制醋、酱,并用曲来治疗消化不良。酶的系统研究起始于19世纪中叶对发酵本质的研究。Pasteur提出,发酵离不了酵母细胞。1897年Buchner成功地用不含细胞的酵母液实现发酵,说明具有发酵作用的物质存在于细胞内,并不依赖活细胞。1926年Sumner首次提取出脲酶,并进行结晶,提出酶的本质是蛋白质。现已有二千余种酶被鉴定出来,其中有二百余种得到结晶,特别是近三十年来,随着蛋白质分离技术的进步,酶的分子结构、酶作用机理的研究得到发展,有些酶的结构和作用机理已被阐明。总之,随着酶学理论不断深入,必将对揭示生命本质研究作出更大的贡献。
第一节 酶的作用特点
酶是生物催化剂(biological catalyst),具有两方面的特性,既有与一般催化剂相同的催化性质,又具有一般催化剂所没有的生物大分子的特征。
酶与一般催化剂一样,只能催化热力学允许的化学反应,缩短达到化学平衡的时间,而不改变平衡点。酶作为催化剂在化学反应的前后没有质和量的改变。微量的酶就能发挥较大的催化作用。酶和一般催化剂的作用机理都是降低反应的活化能(activation energy)。
因为酶是蛋白质,所以酶促反应又固有其特点:
1.高度的催化效率
一般而论,酶促反应速度比非催化反应高107?020倍,例如,反应
H2O2+H2O2→2H2O+O2
在无催化剂时,需活化能18,000卡/克分子;胶体钯存在时,需活化能11,700卡/克分子;有过氧化氢酶(catalase)存在时,仅需活化能2,000卡/克分子以下。
2.高度的专一性
一种酶只作用于一类化合物或一定的化学键,以促进一定的化学变化,并生成一定的产物,这种现象称为酶的特异性或专一性(specificity)。受酶催化的化合物称为该酶的底物或作用物(substrate)。
酶对底物的专一性通常分为以下几种:
(1)绝对特异性(absolutespecifictity)
有的酶只作用于一种底物产生一定的反应,称为绝对专一性,如脲酶(urease),只能催化尿素水解成NH3和CO2,而不能催化甲基尿素水解。
(2)相对特异性(relativespecificity)
一种酶可作用于一类化合物或一种化学键,这种不太严格的专一性称为相对专一性。如脂肪酶(lipase)不仅水解脂肪,也能水解简单的酯类;磷酸酶(phosphatase)对一般的磷酸酯都有作用,无论是甘油的还是一元醇或酚的磷酸酯均可被其水解。
(3)立体异构特异性(stereopecificity)
酶对底物的立体构型的特异要求,称为立体异构专一性或特异性。如α-淀粉酶(α-amylase)只能水解淀粉中α-1,4-糖苷键,不能水解纤维素中的β-1,4-糖苷键;L-乳酸脱氢酶(L-lacticacid dehydrogenase)的底物只能是L型乳酸,而不能是D型乳酸。酶的立体异构特异性表明,酶与底物的结合,至少存在三个结合点。
3.酶活性的可调节性
酶是生物体的组成成份,和体内其他物质一样,不断在体内新陈代谢,酶的催化活性也受多方面的调控。例如,酶的生物合成的诱导和阻遏、酶的化学修饰、抑制物的调节作用、代谢物对酶的反馈调节、酶的别构调节以及神经体液因素的调节等,这些调控保证酶在体内新陈代谢中发挥其恰如其分的催化作用,使生命活动中的种种化学反应都能够有条不紊、协调一致地进行。
4.酶活性的不稳定性
酶是蛋白质,酶促反应要求一定的pH、温度等温和的条件,强酸、强碱、有机溶剂、重金属盐、高温、紫外线、剧烈震荡等任何使蛋白质变性的理化因素都可能使酶变性而失去其催化活性。
第二节 酶的分类和命名
一、酶的分类
国际酶学委员会(I.E.C)规定,按酶促反应的性质,可把酶分成六大类:
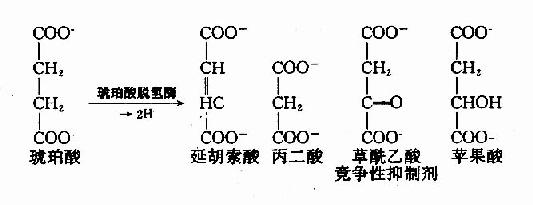
1.氧化还原酶类(oxidoreductases)指催化底物进行氧化还原反应的酶类。例如,乳酸脱氢酶、琥珀酸脱氢酶、细胞色素氧化酶、过氧化氢酶等。
2.转移酶类(transferases)指催化底物之间进行某些基团的转移或交换的酶类。如转甲基酶、转氨酸、己糖激酶、磷酸化酶等。
3.例如、淀粉酶、蛋白酶、脂肪酶、磷酸酶等。
4.裂解酶类(lyases)指催化一个底物分解为两个化合物或两个化合物合成为一个化合物的酶类。例如柠檬酸合成酶、醛缩酶等。
5.异构酶类(isomerases)指催化各种同分异构体之间相互转化的酶类。例如,磷酸丙糖异构酶、消旋酶等。
6.合成酶类(连接酶类,ligases)指催化两分子底物合成为一分子化合物,同时还必须偶联有ATP的磷酸键断裂的酶类。例如,谷氨酰胺合成酶、氨基酸:tRNA连接酶等。
二、酶的命名
(一)习惯命名法
1.一般采用底物加反应类型而命名,如蛋白水解酶、乳酸脱氢酶、磷酸己糖异构酶等。
2.对水解酶类,只要底物名称即可,如蔗糖酶、胆硷酯酶、蛋白酶等。
3.有时在底物名称前冠以酶的来源,如血清谷氨酸-丙酮酸转氨酶、唾液淀粉酶等。
习惯命名法简单,应用历史长,但缺乏系统性,有时出现一酶数名或一名数酶的现象。
(二)系统命名法
鉴于新酶的不断发展和过去文献中对酶命名的混乱,国际酶学委员会规定了一套系统的命名法,使一种酶只有一种名称。它包括酶的系统命名和4个数字分类的酶编号。例如对催化下列反应酶的命名。
ATP+D—葡萄糖→ADP+D—葡萄糖-6-磷酸
该酶的正式系统命名是:ATP:葡萄糖磷酸转移酶,表示该酶催化从ATP中转移一个磷酸到葡萄糖分子上的反应。它的分类数字是:E.C.2.7.1.1,E.C代表按国际酶学委员会规定的命名,第1个数字(2)代表酶的分类名称(转移酶类),第2个数字(7)代表亚类(磷酸转移酶类),第3个数字(1)代表亚亚类(以羟基作为受体的磷酸转移酶类),第4个数字(1)代表该酶在亚-亚类中的排号(D葡萄糖作为磷酸基的受体)。
第三节 酶的分子组成和化学结构
一、酶的分子组成
根据酶的组成成份,可分单纯酶和结合酶两类。
单纯酶(simpleenzyme)是基本组成单位仅为氨基酸的一类酶。它的催化活性仅仅决定于它的蛋白质结构。脲酶、消化道蛋白酶、淀粉酶、酯酶、核糖核酸酶等均属此列。
结合酶(conjugatedenzyme)的催化活性,除蛋白质部分(酶蛋白apoenzyme)外,还需要非蛋白质的物质,即所谓酶的辅助因子(cofactors),两者结合成的复合物称作全酶(holoenzyme),即:
| 全酶 |
=酶 蛋 白 |
+ 辅助因子 |
| (结合蛋白质) |
(蛋白质部分) |
(非蛋白质部分) |
酶的辅助因子可以是金属离子,也可以是小分子有机化合物。常见酶含有的金属离子有K+、Na+、Mg2+、Cu2+、(或Cu+)、Zn2+和Fe2+(或Fe3+)等。它们或者是酶活性的组成部分;或者是连接底物和酶分子的桥梁;或者在稳定酶蛋白分子构象方面所必需。小分子有机化合物是些化学稳定的小分子物质,其主要作用是在反应中传递电子、质子或一些基团,常可按其与酶蛋白结合的紧密程度不同分成辅酶和辅基两大类。辅酶(coenzyme)与酶蛋白结合疏松,可以用透析或超滤方法除去;辅基(prostheticgroup)与酶蛋白结合紧密,不易用透析或超滤方法除去,辅酶和辅基的差别仅仅是它们与酶蛋白结合的牢固程度不同,而无严格的界限。
现知大多数维生素(特别是B族维生素)是组成许多酶的辅酶或辅基的成分(见表2-1)。它们的化学结构式见生物氧化章。体内酶的种类很多,而辅酶(基)的种类却较少,通常一种酶蛋白只能与一种辅酶结合,成为一种特异的酶,但一种辅酶往往能与不同的酶蛋白结合构成许多种特异性酶。酶蛋白在酶促反应中主要起识别底物的作用,酶促反应的特异性、高效率以及酶对一些理化因素的不稳定性均决定于酶蛋白部分。
表2-1 B族维生素及其辅酶形式
| B族维生素 |
辅酶形式 |
主要作用 |
| 硫胺素(B1) |
硫胺素焦磷酸酯(TPP) |
α-酮酸氧化脱羧酮基转换作用 |
| 硫辛酸 |
6,8-二硫辛酸
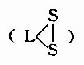 |
α-酮酸氧化脱羧 |
| 泛酸 |
辅酶A(CoA) |
酰基转换作用 |
| 核黄素(B2) |
黄素单核苷酸(FMN) 黄素腺嘌呤二核苷酸(FAD) |
氢原子转移 氢原子转移 |
| 尼克酰胺(PP) |
尼克酰胺腺嘌呤二核苷酸(NAD+) 尼克酰胺腺嘌呤二核苷酸磷酸(NADP+) |
氢原子转移 氢原子转移 |
| 吡哆素(B6) |
磷酸吡哆醛 |
氨基酸代谢 |
| 生物素(H) |
生物素 |
羧化作用 |
| 叶酸 |
四氢叶酸 |
“一碳基团”转移 |
| 钴胺素(B12) |
5-甲基钴铵素 5-脱氧腺苷钴铵素 |
甲基转移 |
二、酶的分子结构和活性中心
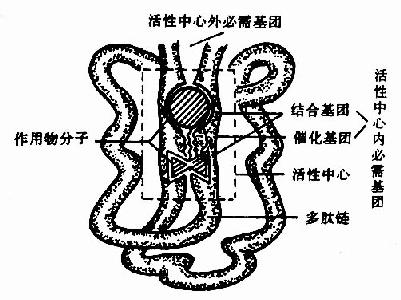
图2-1 酶活性中心示意图
酶的分子中存在有许多功能基团例如,-NH2、-COOH、-SH、-OH等,但并不是这些基团都与酶活性有关。一般将与酶活性有关的基团称为酶的必需基团(essentialgroup)。有些必需基团虽然在一级结构上可能相距很远,但在空间结构上彼此靠近,集中在一起形成具有一定空间结构的区域,该区域与底物相结合并将底物转化为产物,这一区域称为酶的活性中心(active center),对于结合酶来说,辅酶或辅基上的一部分结构往往是活性中心的组成成分。
构成酶活性中心的必需基团可分为两种,与底物结合的必需基团称为结合基团(binding group),促进底物发生化学变化的基团称为催化基团(catalyticgroup)。活性中心中有的必需基团可同时具有这两方面的功能。还有些必需基团虽然不参加酶的活性中心的组成,但为维持酶活性中心应有的空间构象所必需,这些基团是酶的活性中心以外的必需基团。
酶分子很大,其催化作用往往并不需要整个分子,如用氨基肽酶处理木瓜蛋白酶,使其肽链自N端开始逐渐缩短,当其原有的180个氨基酸残基被水解掉120个后,剩余的短肽仍有水解蛋白质的活性。又如将核糖核酸酶肽链C末端的三肽(棻麠丝楃?切断,余下部分也有酶的活性,足见某些酶的催化活性仅与其分子的一小部分有关。
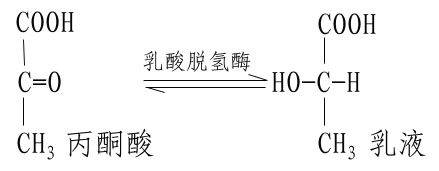
不同的酶有不同的活性中心,故对底物有严格的特异性。例如乳酸脱氢酶是具有立体异构特异性的酶,它能催化乳酸脱氢生成丙酮酸的可逆反应:
L(+)乳酸通过其不对称碳原子上的桟H3、桟OOH及桹H基分别与乳酸脱氢酶活性中心的A、B及C三个功能基团结合,故可受酶催化而转变为丙酮酸。而D(-)乳酸由于桹H、桟OOH的空间位置与L(+)乳酸相反,与酶的三个结合基团不能完全配合,故不能与酶结合受其催化(图2)。由此可见,酶的特异性不但决定于酶活性中心的功能基团的性质,而且还决定于底物和活性中心的空间构象,只有那些有一定的化学结构,能与酶的结合基团结合,而且空间构型又完全适应的化合物,才能作为酶的底物。
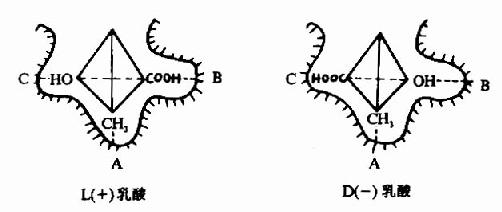
图2-2 乳酸脱氢酶的立体异构特异性
A、B、C分别为LDH活性中心的三个功能基团
但是,酶的结构不是固定不变的,有人提出酶分子(包括辅酶在内)的构型与底物原来并非吻合,当底物分子与酶分子相碰时,可诱导酶分子的构象变得能与底物配合,然后底物才能与酶的活性中心结合,进而引起底物分子发生相应化学变化,此即所谓酶作用的诱导契合学说(induced fit theory)。用X衍射分析的方法已证明,酶在参与催化作用时发生了构象变化。
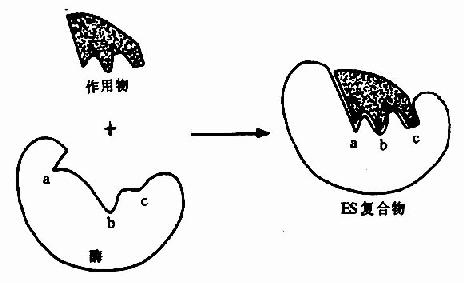
图2-3 底物与酶相互作用的“诱导契合”模式图
第四节 酶的作用机理
一、酶作用在于降低反应活化能
在任何化学反应中,反应物分子必须超过一定的能阈,成为活化的状态,才能发生变化,形成产物。这种提高低能分子达到活化状态的能量,称为活化能。催化剂的作用,主要是降低反应所需的活化能,以致相同的能量能使更多的分子活化,从而加速反应的进行。
酶能显着地降低活化能,故能表现为高度的催化效率(图2?)。例如前述的H2O2酶的例子,可以显着地看出,酶能降低反应活化能,使反应速度增高千百万倍以上。
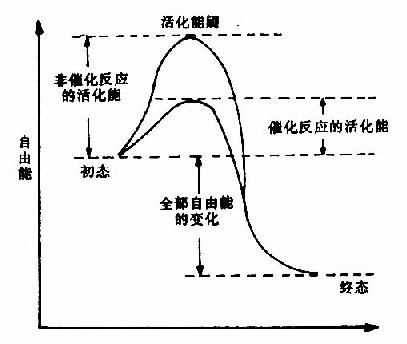
图2-4 非催化过程和催化过程自由能的变化
二、中间复合物学说
目前一般认为,酶催化某一反应时,首先在酶的活性中心与底物结合生成酶-底物复合物,此复合物再进行分解而释放出酶,同时生成一种或数种产物,此过程可用下式表示:
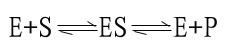
上式中E代表酶,S代表底物,ES代表酶椀孜锔春衔?中间产物),P代表反应产物。由于ES的形成速度很快,且很不稳定,一般不易得到ES复合物存在的直接证据。但从溶菌酶结构的研究中,已制成它与底物形成复合物的结晶,并得到了X线衍射图,证明了ES复合物的存在。
ES的形成,改变了原来反应的途径,可使底物的活化能大大降低,从而使反应加速。
三、酶作用高效率的机理
详细机制仍不太清楚,主要有下列四种因素:
1.趋近效应(approximation)和定向效应(oientation)
酶可以将它的底物结合在它的活性部位由于化学反应速度与反应物浓度成正比,若在反应系统的某一局部区域,底物浓度增高,则反应速度也随之提高,此外,酶与底物间的靠近具有一定的取向,这样反应物分子才被作用,大大增加了ES复合物进入活化状态的机率(图2-5)。
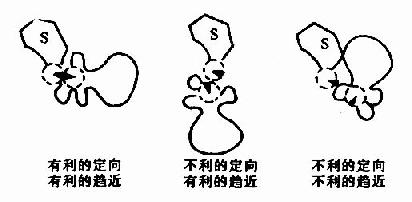
图2-5 底物分子和酶活性中心上的一个催化基团在相互作用时的趋近效应
2.张力作用(distortion or strain)
底物的结合可诱导酶分子构象发生变化,比底物大得多的酶分子的三、四级结构的变化,也可对底物产生张力作用,使底物扭曲,促进ES进入活性状态(图2-6)。

图2-6 酶的活性中心诱导契合使底物分子扭曲
3.酸碱催化作用(acid-base catalysis)
酶的活性中心具有某些氨基酸残基的R基团,这些基团往往是良好的质子供体或受体,在水溶液中这些广义的酸性基团或广义的碱性基团对许多化学反应是有力的催化剂。
| 某些质子供体基团 |
某些受子体基团 |
| -COOH |
-COO- |
| -NH+3 |
-NH2 |
| -SH |
-S- |
 |
4.共价催化作用(covalent catalysis)
某些酶能与底物形成极不稳定的、共价结合的ES复合物,这些复合物比无酶存在时更容易进行化学反应。
例如:无酶催化的反应 RX+H2O→ROH+Hx慢
有酶存在时 RX+E桹H→ROH+EX快
EX+H2O→E桹H+HX快
第五节 酶促反应的动力学
酶促反应动力学(kineticsof enzyme-catalyzed reactions)是研究酶促反应速度及其影响因素的科学。这些因素主要包括酶的浓度、底物的浓度、pH、温度、抑制剂和激活剂等。在研究某一因素对酶促反应速度的影响时,应该维持反应中其它因素不变,而只改变要研究的因素。但必须注意,酶促反应动力学中所指明的速度是反应的初速度,因为此时反应速度与酶的浓度呈正比关系,这样避免了反应产物以及其他因素的影响。
酶促反应动力学的研究有助于阐明酶的结构与功能的关系,也可为酶作用机理的研究提供数据;有助于寻找最有利的反应条件,以最大限度地发挥酶催化反应的高效率;有助于了解酶在代谢中的作用或某些药物作用的机理等,因此对它的研究具有重要的理论意义和实践意义。
一、酶浓度对反应速度的影响
在一定的温度和pH条件下,当底物浓度大大超过酶的浓度时,酶的浓度与反应速度呈正比关系(图2-7)。
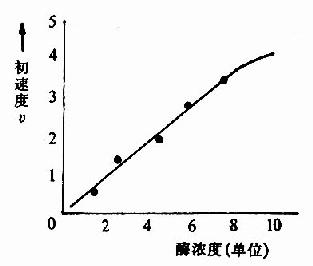
二、底物浓度对反应速度的影响
在酶的浓度不变的情况下,底物浓度对反应速度影响的作用呈现矩形双曲线(rectangular hyperbola)(图2-8)。
 |
 |
| 图2-7 酶浓度对反应初速度的影响 |
图2-8 底物浓度对反应初速度的影响 |

在底物浓度很低时,反应速度随底物浓度的增加而急骤加快,两者呈正比关系,表现为一级反应。随着底物浓度的升高,反应速度不再呈正比例加快,反应速度增加的幅度不断下降。如果继续加大底物浓度,反应速度不再增加,表现为0级反应。此时,无论底物浓度增加多大,反应速度也不再增加,说明酶已被底物所饱和。所有的酶都有饱和现象,只是达到饱和时所需底物浓度各不相同而已。
(一)米曼氏方程式
解释酶促反应中底物浓度和反应速度关系的最合理学说是中间产物学说。酶首先与底物结合生成酶椀孜锔春衔?中间产物),此复合物再分解为产物和游离的酶。
Michaelis和Menten在前人工作的基础上,经过大量的实验,1913年前后提出了反应速度和底物浓度关系的数学方程式,即著名的米椔?戏匠淌?michaelismenten equation).
V=Vmax[S]/Km+[S]
Vmax指该酶促反应的最大速度,[S]为底物浓度,Km是米氏常数,V是在某一底物浓度时相应的反应速度。当底物浓度很低时,[S]《Km,则V≌Vmax/Km[S],反应速度与底物浓度呈正比。当底物浓度很高时,[S]》Km,此时V≌Vmax,反应速度达最大速度,底物浓度再增高也不影响反应速度(图2-9)。
图2-9 酶与不同浓度的底物相互作用模式
(二)米-曼氏方程式的推导
米-曼氏方程式提出后又经riggs和Haldane的充实和发展,经补充和发展的米-曼氏方程工推导如下:
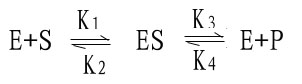
(1)
式中K1、K2、K3、K4分别为各向反应的速度常数。
从式(1)中知,ES的生成途径来自E+S和E+P,但其中E+P生成ES的速度极小(尤其在起始阶段,P的生成很少),可以忽略不计,又因为底物浓度大大超过酶的浓度,[S]》[E],中间产物ES中的S浓度可以忽略不计,因此,ES的生成速度为:
| d[ES] |
= |
K1([Et]-[ES])·[S] |
(2) |
| dt |
其中[Et]-[ES]为游离酶的浓度,ES的分解速度为:
| - |
[ES] |
= |
K2[ES]+K3[ES]=(K2+K3)[ES] |
(3) |
| dt |
当反应体系处于稳态时,ES生成和分解的速度相等,即
K1([Et]-[ES])·[S]=(K2+K3)[ES]
| K2+K3 |
= |
[Et]-[ES] |
·[S] |
| K1 |
[ES] |
令K2+K3/K1=Km 则 Km=[Et]-[ES]/[ES]·[S]
[ES]=[Et][S]/Km+[S] (4)
由于反应速度取决于产物P的生成量,故
V=K3[ES (5)
在酶促反应达最大速度时,所有的酶分子都已与底物结合形成中间产物,此时
[Et]=[ES] (6)
那么 Vmax=K3[Et] (7)
在(4)式两边乘以K3得:
K3·[ES]=K3·[Et][S]/Km+[S] 以(5)和(7)式代入,即:
V=Vmax[S]/Km+[S]
(三)米氏常数的意义
当反应速度为最大速度一半时,米氏方程可以变换如下:
½Vmax=Vmax[S]/Km+[S]
进一步整理可得到:
Km=[S]
可知,Km值等于酶反应速度为最大速度一半时的底物浓度。
因为Km=K2+K3/K1,当K2》K3,即ES解离成E和S的速度大大超过分离成E和P的速度时,K3可以忽略不计,此时Km值近似于ES解离常数KS,此时Km值可用来表示酶对底物的亲和力。
Km=K2/K1=[E][S]/[ES]=KS
Km值愈大,酶与底物的亲和力愈小;Km值愈小,酶与底物亲和力愈大。酶与底物亲和力大,表示不需要很高的底物浓度,便可容易地达到最大反应速度。但是KS值并非在所有酶促反应中都远小于K2,所以Ks值(又称酶促反应的底物常数)和Km值的涵义不同,不能互相代替使用。
Km值是酶的特征性常数,只与酶的性质,酶所催化的底物和酶促反应条件(如温度、pH、有无抑制剂等)有关,与酶的浓度无关。酶的种类不同,Km值不同,同一种酶与不同底物作用时,Km值也不同。各种酶的Km值范围很广,大致在10-1~10-6M之间。
当K3不远远小于K2和K1时,Km表示整个反应的化学平衡的常数。
如果Km值已知,任何底物浓度时酶的饱和度(形成中间产物的酶占总酶的比例,saturation fraction fEs)fEs便可计算出来。
fES=[ES]/[Et]=K3[ES]/K3[Et]=V/Vmax=[S]/Km+[S]
(四)Km和Vmax的求法
如图2?所示,底物浓度曲线是矩形双曲线。
从图中很难精确地测出Km和Vmax。为此人们将米氏方程进行种种变换,将曲线作图转变成直线作图。
1.双倒数作图(double?reciprocal plot or Lineweaver?Burk plot)
将米氏方程两边取倒数,可转化为下列形式:
1/V=Km/Vmax·1/[S]+1/Vmax
从图2-10可知,1/V对1/[S]的作图得一直线,其斜率是Km/V,在纵轴上的截距为1/Vmax,横轴上的截距为-1/Km。此作图除用来求Km和Vmax值外,在研究酶的抑制作用方面还有重要价值。
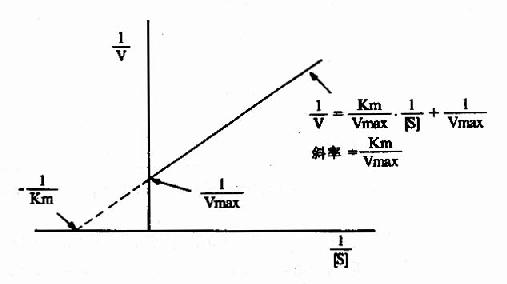
图2-10 双倒数作图法
![v对v/[s]作图法](/__local/3/B1/B7/1C10BD31B82D37C3E8400BD3F81_BEA72BA9_2044.jpg?e=.jpg)
图2-11 v对v/[s]作图法
2.V对V〖〗[S][SX)]法(Eadie?Hofstee plot)
将米氏方程经移项整理后可写成
VKm+V[S]=Vm[S]
V[S]=Vm[S]-VKm
故V=Vm-KmV/[S]
以V为纵坐标对V/[S]横坐标作图,所得直线,其纵轴的截距为Vmax,斜率为Km(图2-11)。
必须指出米氏方程只适用于较为简单的酶作用过程,对于比较复杂的酶促反应过程,如多酶体系、多底物、多产物、多中间物等,还不能全面地籍此概括和说明,必须借助于复杂的计算过程。
三、pH对反应速度的影响
酶反应介质的pH可影响酶分子,特别是活性中心上必需基团的解离程度和催化基团中质子供体或质子受体所需的离子化状态,也可影响底物和辅酶的解离程度,从而影响酶与底物的结合。只有在特定的pH条件下,酶、底物和辅酶的解离情况,最适宜于它们互相结合,并发生催化作用,使酶促反应速度达最大值,这种pH值称为酶的最适pH(optimum pH)。它和酶的最稳定pH不一定相同,和体内环境的pH也未必相同。
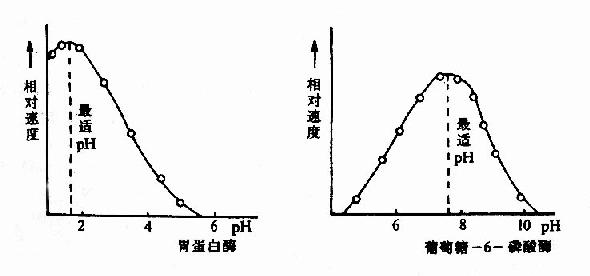
图2-12 胃蛋白酶和葡萄糖-6-磷酸酶的pH活性曲线
动物体内多数酶的最适pH值接近中性,但也有例外,如胃蛋白酶的最适pH约1.8,肝精氨酸酶最适pH约为9.8(见表2-2)。
表2-2 一些酶的最适pH
| 酶 |
最适pH |
酶 |
最适pH |
酶 |
最适pH |
| 胃蛋白酶 |
1.8 |
过氧化氢酶 |
7.6 |
延胡索酸酶 |
7.8 |
| 胰蛋白酶 |
7.7 |
精氨酸酶 |
9.8 |
核糖核酸酶 |
7.8 |
最适pH不是酶的特征性常数,它受底物浓度、缓冲液的种类和浓度以及酶的纯度等因素的影响。
溶液的pH值高于和低于最适pH时都会使酶的活性降低,远离最适pH值时甚至导致酶的变性失活。测定酶的活性时,应选用适宜的缓冲液,以保持酶活性的相对恒定。
四、温度对反应速度的影响
化学反应的速度随温度增高而加快。但酶是蛋白质,可随温度的升高而变性。在温度较低时,前一影响较大,反应速度随温度升高而加快,一般地说,温度每升高10℃,反应速度大约增加一倍。但温度超过一定数值后,酶受热变性的因素占优势,反应速度反而随温度上升而减缓,形成倒V形或倒U形曲线。在此曲线顶点所代表的温度,反应速度最大,称为酶的最适温度(optimum temperature)(图2-13)。
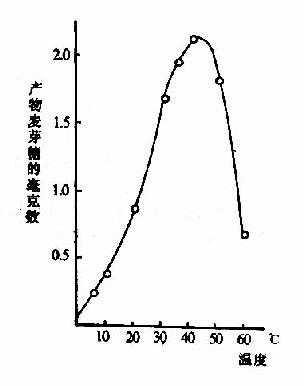
图2-13 温度对唾液淀粉酶活性影响
从动物组织提取的酶,其最适温度多在35℃~40℃之间,温度升高到60℃以上时,大多数酶开始变性,80℃以上,多数酶的变性不可逆。酶的活性虽然随温度的下降而降低,但低温一般不破坏酶。温度回升后,酶又恢复活性。临床上低温麻醉就是利用酶的这一性质以减慢组织细胞代谢速度,提高机体对氧和营养物质缺乏的耐受体,有利于进行手术治疗。
酶的最适温度不是酶的特征性常数,这是因为它与反应所需时间有关,不是一个固定的值。酶可以在短时间内耐受较高的温度,相反,延长反应时间,最适温度便降低。
五、抑制剂对反应速度的影响
凡能使酶的活性下降而不引起酶蛋白变性的物质称做酶的抑制剂(inhibitor)。使酶变性失活(称为酶的钝化)的因素如强酸、强碱等,不属于抑制剂。通常抑制作用分为可逆性抑制和不可逆性抑制两类。
(一)不可逆性抑制作用(irreversibleinhibition)
不可逆性抑制作用的抑制剂,通常以共价键方式与酶的必需基团进行不可逆结合而使酶丧失活性,按其作用特点,又有专一性及非专一性之分。
1.非专一性不可逆抑制
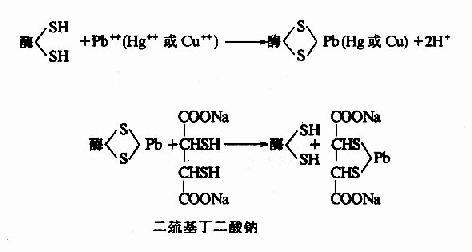
抑制剂与酶分子中一类或几类基团作用,不论是必需基团与否,皆可共价结合,由于其中必需基团也被抑制剂结合,从而导致酶的失活。某些重金属(Pb++、Cu++、Hg++)及对氯汞苯甲酸等,能与酶分子的巯基进行不可逆适合,许多以巯基作为必需基团的酶(通称巯基酶),会因此而遭受抑制,属于此种类型。用二巯基丙醇(british anti?lewisite,BAL)或二巯基丁二酸钠等含巯基的化合物可使酶复活。
2.专一性不可逆抑制
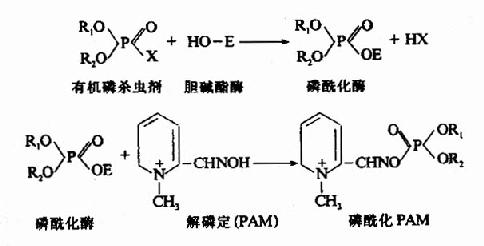
此属抑制剂专一地作用于酶的活性中心或其必需基团,进行共价结合,从而抑制酶的活性。有机磷杀虫剂能专一作用于胆碱酯酶活性中心的丝氨酸残基,使其磷酰化而不可逆抑制酶的活性。当胆碱酯酶被有机磷杀虫剂抑制后,胆碱能神经末稍分泌的乙酰胆碱不能及时分解,过多的乙酰胆碱会导致胆碱能神经过度兴奋的症状。解磷定等药物可与有机磷杀虫剂结合,使酶和有机磷杀虫剂分离而复活。
(二)可逆性抑制(reversible inhibition)
抑制剂与酶以非共价键结合,在用透析等物理方法除去抑制剂后,酶的活性能恢复,即抑制剂与酶的结合是可逆的。这类抑制剂大致可分为以下二类。
1.竞争性抑制(competitive inhibition)
(1)含义和反应式
抑制剂I和底物S对游离酶E的结合有竞争作用,互相排斥,已结合底物的ES复合体,不能再结合I。同样已结合抑制剂的EI复合体,不能再结合S。
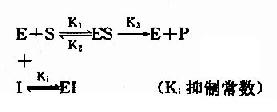
抑制剂I在化学结构上与底物S个相似,能与底物S竞争酶E分子活性中心的结合基团,因此,抑制作用大小取决于抑制剂与底物的浓度比,加大底物浓度,可使抑制作用减弱。
例如,丙二酸、苹果酸及草酰乙酸皆和琥珀酸的结构相似,是琥珀酸脱氢酶的竞争性抑制剂。
(2)反应速度公式及作图
按米氏公式推导方法,也可演算出竞争性抑制时,抑制剂、底物和反应速度之间的动力学关系及其双倒数方程式为:
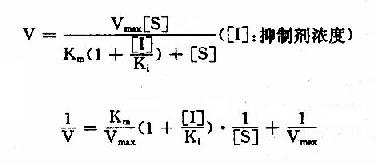
以1V分别为横坐标和纵坐标作图,此方程式可绘成竞争性抑制作用的特性曲线(图2-14)。
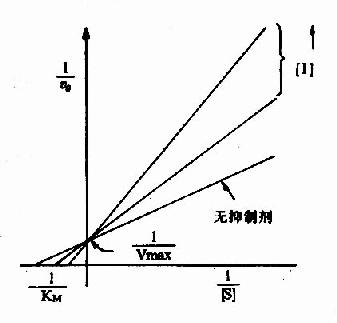
图 2-14 竞争性抑制
有竞争性抑制剂存在的曲线与无抑制剂的曲线相交于纵坐标I/Vmax处,但横坐标的截距,因竞争性抑制存在变小,说明该抑制作用,并不影响酶促反应的最大速度,而使Km值变大。
很多药物都是酶的竞争性抑制剂。例如磺胺药与对氨基苯甲酸具有类似的结构(如图2-15),而对氨基苯甲酸、二氢喋呤及谷氨酸是某些细菌合成二氢叶酸的原料,后者能转变为四氢叶酸,它是细菌合成核酸不可缺少的辅酶。由于磺胺药是二氢叶酸合成酶的竞争性抑制剂,进而减少菌体内四氢叶酸的合成,使核酸合成障碍,导致细菌死亡。抗菌增效剂-甲氧苄氨嘧啶(TMP)能特异地抑制细菌的二氢叶酸还原为四氢叶酸,故能增强磺胺药的作用。
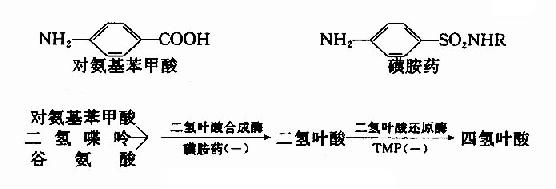
图2-15 磺胺药物的抑菌作用
2.非竞争性抑制(non-competitive inhibition)
(1)含义和反应式
抑制剂I和底物S与酶E的结合完全互不相关,既不排斥,也不促进结合,抑制剂I可以和酶E结合生成EI,也可以和ES复合物结合生成ESI。底物S和酶E结合成ES后,仍可与I结合生成ESI,但一旦形成ESI复合物,再不能释放形成产物P。
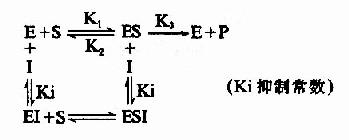
I和S在结构上一般无相似之处,I常与酶分子上结合基团以外的化学基团结合,这种结合并不影响底物和酶的结合,增加底物浓度并不能减少I对酶的抑制程度。
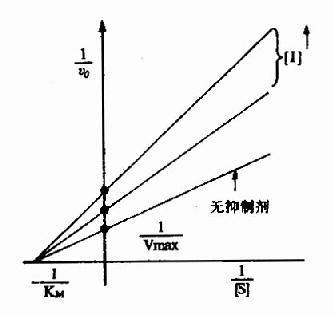
图2-16 非竞争性抑制
(2)反应速度公式及作图
按米氏公式推导方法可演算出非竞争性抑制时,抑制剂、底物浓度和反应速度之间动力学关系:
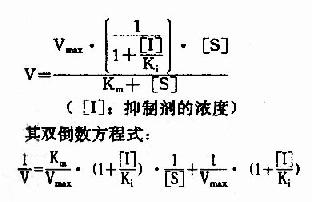
以1V分别为横坐标和纵坐标作图,此方程式可绘成非竞争性抑制作用的特性曲线(图2-16)。
有非竞争性抑制剂存在的曲线与无抑制剂存在的曲线相交于横坐标-1/Km处,纵坐标截距,因非竞争性抑制剂的存在而变大,说明该抑制作用,并不影响底物与酶的亲和力,而使酶促最大反应速度变小。
如赖氨酸是精氨酸酶的竞争性抑制剂,而中性氨基酸(如丙氨酸)则是非竞争性抑制剂。
总上所述,酶的竞争性和非竞争性抑制可通过双倒数作图加以区别。Vmax不因竞争性抑制剂的存在而改变,Km则不因非竞争性抑制剂的存在而改变。
六、激活剂对酶促反应速度的影响
能使酶活性提高的物质,都称为激活剂(activator),其中大部分是离子或简单的有机化合物。如Mg++是多种激酶和合成酶的激活剂,动物唾液中的α-淀粉酶则受Cl-的激活。
第六节 酶在体内存在的几种主要形式
一、酶原
有些酶在细胞内合成时,或初分泌时,没有催化活性,这种无活性状态的酶的前身物称为酶原(zymogen)。酶原向活性的酶转化的过程称为酶原的激活。酶原激活实际上是酶的活性中心形成或暴露的过程。
胃蛋白酶、胰蛋白酶、胰糜蛋白酶、羧基肽酶、弹性蛋白酶在它们初分泌时都是以无活性的酶原形式存在,在一定条件下(表2?)才转化成相应的酶。
表2-3 某些酶原的激活过程
| 酶原 |
激活条件 |
活化的酶 |
|
水解掉的肽段 |
| 胃蛋白酶原 |
 |
胃蛋白酶 |
+ |
六个多肽片段 |
| 胰蛋白酶原 |
 |
胰蛋白酶 |
+ |
六肽 |
| 糜蛋白酶原A |
 |
α-糜蛋白酶 |
+ |
两个二肽 |
| 羧基肽酶原A |
 |
羧基肽酶A |
+ |
几个碎片 |
| 弹性蛋白酶原 |
|
弹性蛋白酶 |
+ |
几个碎片 |
例如,胰蛋白酶原进入小肠后,受肠激酶或胰蛋白酶本身的激活,第6位赖氨酸与第7位异亮氨酸残基之间的肽键被切断,水解掉一个六肽,酶分子空间构象发生改变,产生酶的活性中心,于是胰蛋白酶原变成了有活性的胰蛋白酶(图2-17)。
除消化道的蛋白酶外,血液中有关凝血和纤维蛋白溶解的酶类,也都以酶原的形式存在。
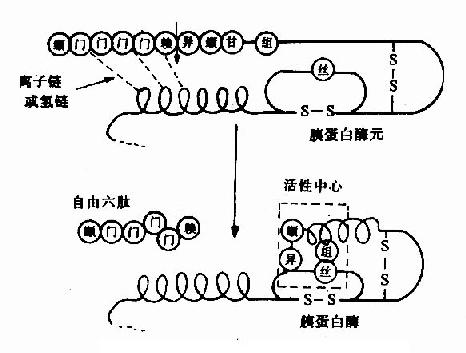
图2-17 胰蛋白酶原激活示意图
酶原激活的生理意义在于避免细胞内产生的蛋白酶对细胞进行自身消化,并可使酶在特定的部位和环境中发挥作用,保证体内代谢的正常进行。
二、同工酶
同工酶(isoenzyme)是指催化的化学反应相同,酶蛋白的分子结构、理化性质乃至免疫学性质不同的一组酶。这类酶存在于生物的同一种属或同一个体的不同组织、甚至同一组织或细胞中。
现已发现有数种同工酶。如6?磷酸葡萄糖脱氢酶、乳酸脱氢酶、酸性和碱性磷酸酶、谷丙转氨酶和谷草转氨酸、肌酸磷酸激酶、核糖核酸酶、过氧化酶和胆碱酯酶等。其中乳酸脱氢酶最为大家所熟悉,乳酸脱氢酶(LDH)有五种同工酶,它们的分子量在130,000~150,000范围内,都由四个亚基组成。LDH的亚基可以分为两型:骨骼肌型(M型)和心肌型(H型)。M、H亚基的氨基酸组成有差别,可用电泳分离。其免疫抗体无交叉反应。两种亚基以不同比例组成五种四聚体即为一组LDH同工酶LDH1(H4)、LDH2(H3M)、LDH3(H2M2)、LDH4(HM3)和LDH5(M4)。电泳时都移向正极,其速度以LDH1为最快,依次递减,以LDH5为最慢。若用12M尿素或5M盐酸胍溶液处理,M亚基和H亚基可以分开,但此时LDH无酶的活性。
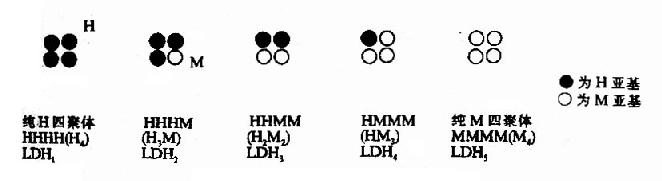
图2-18 LDH同工酶结构模式图
LDH同工酶的两种不同肽链是受不同基因控制产生的。不同类型的LDH同工酶在不同

图2-19人体某些组织中乳酸脱氢酶同工酶电泳示意图
组织中的比例不同(图2?9),心肌中以LDH1及LDH2较为丰富,骨骼肌及肝中含LDH5及LDH4较多。这都与它们的生理功能关。LDH1和LDH2对乳酸亲和力高,易使乳酸脱氢氧化生成丙酮酸,后者进一步氧化可释放出能量供心肌活动的需要;LDH5与LDH4对丙酮酸的亲和力高,而使它得氢还原成乳酸,这对保证肌肉在短暂缺氧时仍可获得能量有关(见糖代谢章)。
在临床检验方面,通过观测病人血清中LDH同工酶的电泳图谱,辅助诊断哪些器官组织发生病变,这远较单纯测定血清LDH总活性的方法敏感。例如,心肌受损病人血清LDH1含量上升,肝细胞受损者血清LDH5含量增高。
三、变构酶
1.概念
有些酶除了活性中心外,还有一个或几个部位,当特异性分子非共价地结合到这些部位时,可改变酶的构象,进而改变酶的活性,酶的这种调节作用称为变构调节(allosteric regulation),受变构调节的酶称变构酶(allostericenzyme),这些特异性分子称为效应剂(effector)。变构酶分子组成,一般是多亚基的,分子中凡与底物分子相结合的部位称为催化部位(catalytic site),凡与效应剂相结合的部位称为调节部位(regulatorysite),这二部位可以在不同的亚基上,或者位于同一亚基。
2.机理
(1)一般变构酶分子上有二个以上的底物结合位点。当底物与一个亚基上的活性中心结合后,通过构象的改变,可增强其他亚基的活性中心与底物的结合,出现正协同效应(positivecooperative effect)。使其底物浓度曲线呈S形。即底物浓度低时,酶活性的增加较慢,底物浓度高到一定程度后,酶活性显著加强,最终达到最大值Vmax(图2-20)。
多数情况下,底物对其变构酶的作用都表现正协同效应,但有时,一个底物与一个亚基的活性中心结合后,可降低其他亚基的活性中心与底物的结合,表现负协同效应(negative cooperative effect)。如3-磷酸甘油醛脱氢酶对NAD+的结合为负协同效应。
(2)变构酶除活性中心外,存在着能与效应剂作用的亚基或部位,称调节亚基(或部位),效应剂与调节亚基以非共价键特异结合,可以改变调节亚基的构象,进而改变催化亚基的构象,从而改变酶活性。凡使酶活性增强的效应剂称变构激活剂(allosteric activitor),它能使上述S型曲线左移,饱和量的变构激活剂可将S形曲线转变为矩形双曲线(图2?0)。凡使酶活性减弱的效应剂称变构抑制剂(allosteric inhibitor),能使S形曲线右移。例如,ATP是磷酸果糖激酶的变构抑制剂,而ADP、AMP为其变构激活剂。
(3)由于变构酶动力学不符合米-曼氏酶的动力学,所以当反应速度达到最大速度一半时的底物的浓度,不能用Km表示,而代之以K0.55表示(图2-20)。为了解释变构酶协同效应的机制并推导出动力学曲线方程式,不少人曾提出各种模型,各有优缺点,现将有关变构作用的Hill模式内容附本章节后,供学习参考。?
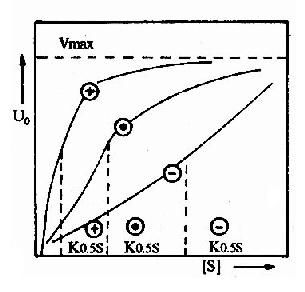
图2-20 变构酶的底物活性曲线
⊙不加变构剂
加变构抑制剂
3.生理意义
(1)在变构酶的S形曲线中段,底物浓度稍有降低,酶的活性明显下降,多酶体系催化的代谢通路可因此而被关闭;反之,底物浓度稍有升高,则酶活性迅速上升,代谢通路又被打开,因此可以快速调节细胞内底物浓度和代谢速度。
(2)变构抑制剂常是代谢通路的终产物,变构酶常处于代谢通路的开端,通过反馈抑制,可以及早地调节整个代谢通路,减少不必要的底物消耗。
例如葡萄糖的氧化分解可提供能量使AMP、ADP转变成ATP,当ATP过多时,通过变构调节酶的活性,可限制葡萄糖的分解,而ADP、AMP增多时,则可促进糖的分解。随时调节ATP/ADP的水平,可以维持细胞内能量的正常供应。
四、修饰酶
体内有些酶可在其他酶的作用下,将酶的结构进行共价修饰,使该酶活性发生改变,这种调节称为共价修饰调节(covalent modification regulation),这类酶称为修饰酶(prosessingenzyme)。
例如某些酶的巯基发生可逆的氧化还原,一些酶以共价键与磷酸、腺苷等基团的可逆结合,都会引起酶结构的变化而呈现不同的活性。酶的共价修饰是体内代谢调节的另一重要的方式。
五、多酶复合体
多酶复合体(multienzymecomplex)常包括三个或三个以上的酶,组成一个有一定构型的复合体。复合体中第一个酶催化的产物,直接由邻近下一个酶催化,第二个酶催化的产物又为复合体第三酶的底物,如此形成一条结构紧密的“流水生产线”,使催化效率显着提高。葡萄糖氧化分解过程的丙酮酸脱氢酶复合体,属于多酶复合体(详见糖代谢章)。
参考资料
别构机制的模式
为了解释别构酶协同效应的机制并推导出动力学曲线的方程式,不少人曾提出过各种模式,各有优缺点,现在主要把Hill模式叙述如下:
Hill模式
在协同结合模式中最早的一种是Hill在1909年提出的,企图解释氧结合至血红蛋白的S形饱和曲线,现称为Hill模式,后来经Atkinson应用于别构酶反应,他设想在这个系统中,n分子的配体(S)能够一步结合到酶上去:
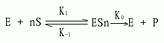
即此反应的总解离常数(K's)由下式决定
而酶的饱和分数
| YS=每分子酶蛋白上已结合的底物分子数/每分子酶蛋白上底物结合位点的总数 |
(6-8) |
又因总的酶浓度[E0]=[E]+[ES0]
| 故 YS=[ESn]/[E0]=[ESn]/[ESn]+[E] |
(6-9) |
合并式6-7和式6-9,消去[ESn],则
| YS=[S]n/K'S+[S]n |
(6-10) |
| YSK'S+YS[S]n=[S]n, |
|
| YSK'S=(1-Y)[S]n |
(6-11) |
| Ys/1-Ys=[S]n/K'S |
(6-11) |
| log |
YS |
=nlog[S]-logK'S |
(6-13) |
| 1-YS |
因此以
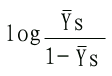
对log[S]作图的话,可得斜率为n,纵轴截距为-logK'S的直线(见下图)。
因v=k0[ESu],Vm=k0[E0],故
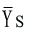 =[ESu]/[E0]=v/vω |
(6-14) |
将式6-10代入式6-14,即得
| [S]n/K'S+[S]n=v/Vm |
|
| Vm[S]n=K'Sv+v[S]n |
(6-15) |
| (Vm-v)[S]n=K'Sv |
(6-16) |
| v/Vm-v=[S]n/K'S |
(6-17) |
| logv/Vm-v=nlog[S]-logK'S |
(6-18) |
式6-13或6-18即为Hill方程式,式6-18如以logv/Vm-v对log[S]作图,也可得一直线(见下图)。
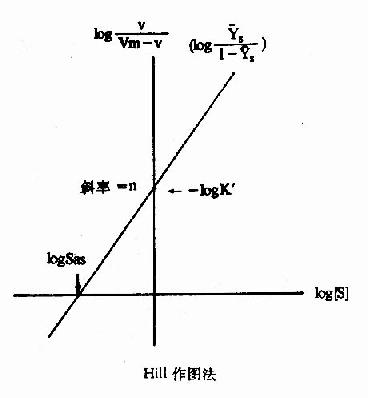
Hill作图法
如v=Vm/2时,式6-19为log1=nlog[S]-logK'S=0(6-19)
此时的[S]即S0.5,故nlogS0.5=logK'5(6-20)式6-18所得的直线斜率为n,纵轴截距为-logk'S,而横轴截距为logK'S/n,即log[S]0.5,但[S0.5]也可在已知logK'S后通过式6?0求取。
上节已述及,S0-5就相当于米曼氏动力学中的Km,当K0《k-1/k1时,可反映别构酶对底物的亲和力,S0.5愈小,亲和力愈大,而K's实际上已与亲和力关系不大,因受到n的影响。故反映底物亲和力的参数,已从非别构酶的Km一项移到别构酶的[S]一项,并且式6?0可看出K'S是随[S]而改变的,不是一个常数。由于K'S的测定是假设V=(1/2)Vm或[S]=S0.5的条件下计算的,故有些作者用S0.5S,来代表别构酶的K'5,以免与Km混淆。
Hill作图法的斜率n,称为Hill系数,即前述的协同系数,一般可用nH或h代表。当nH=1时,式6-1变为V=Vm[S]/(K1+[S]),即米曼氏方程式,表示无协同作用,此时K'或S0.5S,=S0-5=Km,至于nH>1为正协同,nH<1为负协同。
Hill模式比较简单,式6-1或式6-10都是S形曲线方程式,但有不少缺点:(1)按理,Hill系数应等于酶分子中可能有结合底物的位点数,但因忽略了ESn-1,ESn-2…ES1等中间形式的酶底物复合体,根据Hill氏作图计算出来的n值一般均低于真实的位点数。以别构蛋白Hb为例,理论上每分子Hb可结合四分子氧,即n=4,但计算结果n=2.6~2.8。在负协同效应时,每分子酶也结合n个底物(n>1)但计算结果却是n<1。故Hill系数已不能代表结合底物的位点数,而只能作为底物协同性的指标。(2)在S浓度过高(酶90%以上被S饱和)或过低(酶仅10%以下被S饱和)时,Hill线的斜率n常等于1,故当测定别构酶活力时,[S]的范围较广,得出的Hill线不是直线而是折线(见下图)。(3)n分子的底物同时和酶作用,反应的级数为n+1,如n-4则为五级反应,这在动力学上是不可能的。但尽管如此,Hill作图法仍不失是一个求取别构酶S?0.5和鉴定协同类型及协同作用大小的常用方法。
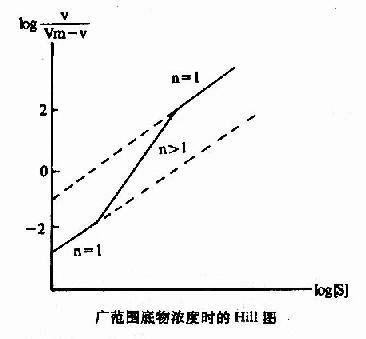
广范围底物浓度时的Hill图

 中医世家
中医世家 浦 标 网
浦 标 网 河南大学精品课程
河南大学精品课程 图书资料室
图书资料室