第一节 药物体内过程
第二节 体内药量变化的时间过程
第三节 药物消除动力学
药物代谢动力学,简称为药动学,研究药物体内过程及体内药物浓度随时间变化的规律。药物在体内虽然不一定集中分布于靶器官,但在分布达到平衡后药理效应强弱与药物血浆浓度成比例。医生可以利用药动学规律科学地计算药物剂量以达到所需的血药浓度并掌握药效的强弱久暂。这样可以比单凭经验处方取得较好的临床疗效。
一、吸 收
药物的吸收(absorption)是指药物自体外或给药部位经过细胞组成的屏蔽膜进入血液循环的过程。多数药物按简单扩散(simple diffusion)物理机制进入体内。扩散速度除取决于膜的性质,面积及膜两侧的浓度梯度外,还与药物的性质有关。分子量小的(200D以下),脂溶性大的(油水分布系数大的),极性小的(不易离子化的)药物较易通过。药物多是弱酸性或弱碱性有机化合物,其离子化程度受其pKa(酸性药物解离常数的负对数值)及其所在溶液的pH而定,这是影响药物跨膜被动转运,吸收分布排泄的一个可变因素。按Handerson-Hasselbalch公式:
由此可见不论弱酸性或弱碱性药物的pKa都是该药在溶液中50%离子化时的pH值,各药有其固定的pKa值。当Pka与pH的差值以数学值增减时,药物的离子型与非离子型浓度比值以指数值相应变化。非离子型药物可以自由穿透,而离子型药物就被限制在膜的一侧,这种现象称为离子障(ion trapping)。例如弱酸性药物在胃液中非离子型多,在胃中即可被吸收。弱碱性药物在酸性胃液中离子型多,主要在小肠吸收。碱性较强的药物如胍乙啶(pKa=11.4)及酸性较强的药物如色甘酸钠(pKa=2.0)在胃肠道基本都已离子化,由于离子障原因,吸收均较难。pKa小于4的弱碱性药物如安定(pKa=3.3)及pKa大于7.5的弱酸性药物如异戊巴比妥(pKa=7.9)在胃肠道pH范围内基本都是非离子型,吸收都快而完全。
少数与正常代谢物相似的药物,如5-氟尿嘧啶、甲基多巴等的吸收是靠细胞中的载体主动转运(active transport)而吸收的,这一主动转运机制对药物在体内分布及肾排泄关系比较密切。易化扩散(facilitated diffusion)是靠载体顺浓度梯度跨膜转运方式,如葡萄糖的吸收,吸收速度较快。固体药物不能吸收,片剂、胶囊剂在胃肠道必须先崩解(disintegration)、溶解(dissolution)后才可能被吸收。
1.胃肠道给药 口服(per os)给药是最常用的给药途径。小肠内pH接近中性,粘膜吸收面广,缓慢蠕动增加药物与粘膜接触机会,是主要吸收部位。药物吸收后通过门静脉进入肝脏。有些药物首次通过肝脏就发生转化,减少进入体循环量,叫做首关消除(first pass elimination)。多数药物口服虽然方便有效,但其缺点是吸收较慢,欠完全,不适用于在胃肠破坏的,对胃刺激大的,首关消除多的药物,也不适用于昏迷及婴儿等不能口服的病人。舌下(sublingual)及直肠(per rectum)给药虽可避免首关消除,吸收也较迅速,但吸收不规则,较少应用。
2.注射给药 静脉注射(intravenous,iv)可使药物迅速而准确地进入体循环,没有吸收过程。肌肉注射(intramuscular,im)及皮下注射(subcutaneous,sc)药物也可全部吸收,一般较口服快。吸收速度取决于局部循环,局部热敷或按摩可加速吸收,注射液中加入少量缩血管药则可延长药物的局部作用。动脉注射(intra-arterial,ia)可将药物输送至该动脉分布部位发挥局部疗效以减少全身反应。例如将溶纤药直接用导管注入冠状动脉以治疗心肌梗塞。注射给药还可将药物注射至身体任何部位发挥作用,如局部麻醉。注射给药需要医护进行,不方便,如果计算剂量有误,过量注入将无法回收。
3.呼吸道给药 肺泡表面积大(达200m2),与血液只隔肺泡上皮及毛细管内皮各一层,而且血流量大,药物只要能到达肺泡,吸收极其迅速,气体及挥发性药物(如全身麻醉药)可直接进入肺泡。药物溶液需要经喷雾器分散为微粒,气雾剂(aerosol)可将药液雾化为直径达5μm左右微粒,可以达到肺泡而迅速吸收,如在雾化器及口鼻罩间加用一个气室则效果更好。2~5μm直径以下的微粒可重被呼出,10μm直径微粒可在小支气管沉积。后者可用于异丙肾上腺素治疗支气管哮喘。较大雾粒的喷雾剂(nebula)只能用于鼻咽部的局部治疗,如抗菌、消炎、祛痰、通鼻塞等。
4.经皮(transdermal)给药 除汗腺外,皮肤不透水,但脂溶性药物可以缓慢通透。许多杀虫药可以经皮吸收中毒。利用这一原理可以经皮给药以达到局部或全身药效,近年来有许多促皮吸收剂加氮酮(azone),可与药物制成贴皮剂,如硝苯地平贴皮剂以达到持久的全身疗效,对于容易经皮吸收的硝酸甘油也可制成缓释贴皮剂预防心绞痛发作,每日只贴一次。
二、分 布
药物进入循环后首先与血浆蛋白结合(plasma protein binding)。酸性药物多与清蛋白结合,碱性药物多与α1酸性糖蛋白结合,还有少数药物与球蛋白结合。这种结合和药物与受体蛋白结合情况相似:
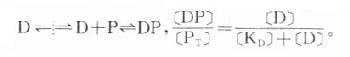
可见药物的血浆蛋白结合量([DP])受药物浓度([D]),血浆蛋白(P)的质和量及解离常数(KD)影响,各药不同而且结合率(血中与蛋白结合的药物与总药量的比值)随剂量增大而减少。药理学书籍收载药物的血浆蛋白结合率是在常用剂量范围内对正常人测定的数值。药物与血浆蛋白的结合是可逆性的,结合后药理活性暂时消失,结合物分子变大不能通过毛细管壁暂时“储存”于血液中。上述反应式中纵向虚线代表毛细管壁,在吸收过程中游离药物穿透毛细管壁进血液后与血浆蛋白结合,反应平衡向右移,有利于吸收。在消除过程中(如肝摄取及肾小管分泌),血中游离药物被除去,反应平衡左移,有利于消除。药物与血浆蛋白结合特异性低,而血浆蛋白结合点有限,两个药物可能竞争与同一蛋白结合而发生置换现象。如某药结合率达99%,当被另药置换而下降1%时,则游离型(具有药理活性)药物浓度在理论上将增加100%,可能导致中毒。但一般药物在被置换过程中,游离型药物会加速被消除,血浆中游离型药物浓度难以持续增高。药物也可能与内源性代谢物竞争与血浆蛋白结合,例如磺胺药置换胆红素与血浆蛋白结合,在新生儿可能导致核黄疸症。血浆蛋白过少(如肝硬化)或变质(如尿毒症)时药物血浆蛋白结合率下降,也容易发生毒性反应。
吸收的药物通过循环迅速向全身组织输送,首先向血流量大的器官分布(distribution),然后向血流量小的组织转移,这种现象称为再分布(redistribution),如硫喷妥先在血流量大的脑中发挥麻醉效应,然后向脂肪等组织转移,效应很快消失。经过一段时间后血药浓度趋向“稳定”,分布达到“平衡”,但各组织中药物并不均等,血浆药物浓度与组织内浓度也不相等。这是由于药物与组织蛋白亲和力不同所致。因此这种“平衡”称为假平衡(pseudoequilibrium),这时血浆药物浓度高低可以反映靶器官药物结合量多少。药物在靶器官浓度决定药物效应强弱,故测定血浆药物浓度可以估算药物效应强度。某些药物可以分布至脂肪、骨质等无生理活性组织形成储库,或结合于毛发指(趾)甲组织。药物的pKa及体液pH是决定药物分布的另一因素,细胞内液 pH(约为7.0)略低于细胞外液(约7.4),弱碱性药物在细胞内浓度略高,弱酸性药物在细胞外液浓度略高,根据这一原理,弱酸性药物苯巴比妥中毒时用碳酸氢钠碱化血液及尿液可使脑细胞中药物向血浆转移并加速自尿排泄,是重要救治措施之一。
血脑屏障(blood-brain barrier) 脑是血流量较大的器官,但药物在脑组织浓度一般较低,这是由于血脑屏障所致。在组织学上血脑屏障是由血-脑、血-脑脊液及脑脊液-脑三种屏障的总称,实际上能阻碍药物穿透的主要是前二者。脑毛细血管内皮细胞间紧密联接,基底膜外还有一层星状细胞包围,药物较难穿透。脑脊液不含蛋白质,即使少量未与血浆蛋白结合的脂溶性药物可以穿透进入脑脊液,其后药物进入静脉的速度较快,故脑脊液中药物浓度总是低于血浆浓度,这是大脑自我保护机制。治疗脑病可以选用极性低的脂溶性药物,例如磺胺药中的磺胺嘧啶。为了减少中枢神经不良反应,对于生物碱可将之季铵化以增加其极性,例如将阿托品季铵化变为甲基阿托品后不能通过血脑屏障,即不致发生中枢兴奋反应。
胎盘屏障(placenta barrier)是胎盘绒毛与子宫血窦间的屏障,由于母亲与胎儿间交换营养成分与代谢废物的需要,其通透性与一般毛细管无显著差别,只是到达胎盘的母体血流量少,进入胎儿循环慢一些罢了。例如母亲注射磺胺嘧啶2小时后才能与胎儿达到平衡。利用这一原理可以在预期胎儿娩出前短时内注射镇静镇痛药,新生儿不致遭受影响。应该注意的是几乎所有药物都能穿透胎盘屏障进入胚胎循环,在妊娠期间应禁用对胎儿发育有影响的药物。
三、生物转化
药物,作为外来活性物质(xenobiotic),机体首先要将之灭活,同时还要促其自体内消除。能大量吸收进入体内的药物多是极性低的脂溶性药物,在排泄过程中易被再吸收,不易消除。体内药物主要在肝脏生物转化(biotransformation) 而失去药理活性,并转化为极性高的水溶性代谢物而利于排出体外。生物转化与排泄统称为消除(elimination)。
生物转化分两步进行,第一步为氧化、还原或水解,第二步为结合。第一步反应使多数药物灭活,但少数例外反而活化,故生物转化不能称为解毒过程。第二步与体内物质结合后总是使药物活性降低或灭活并使极性增加。各药在体内转化过程不同,有的只经一步转化,有的完全不变自肾排出,有的经多步转化生成多个代谢产物。
肝脏微粒体的细胞色素P-450酶系统是促进药物生物转化的主要酶系统,故又简称肝药酶,现已分离出70余种。此酶系统的基本作用是从辅酶Ⅱ及细胞色素 b5 获得两个H+,另外接受一个氧分子,其中一个氧原子使药物羟化,另一个氧原子与两个H+结合成水(RH+NADPH+O2+2H+ →ROH+NADP+ +H2O),没有相应的还原产物,故又名单加氧酶,能对数百种药物起反应(图3-1)。此酶系统活性有限,在药物间容易发生竞争性抑制。它又不稳定,个体差异大,且易受药物的诱导或抑制。例如苯巴比妥能促进光面肌浆网增生,其中P-450酶系统活性增加,加速药物生物转化,这是其自身耐受性及与其他药物交叉耐受性的原因。西米替丁抑制P-450酶系统活性,可使其他药物效应敏化。该酶系统在缺氧条件下可对偶氮及芳香硝基化合物产生还原反应,生成胺基(图3-2)。微粒体内还存在水解酶及葡萄糖醛酸转移酶。
图3-1 细胞色素P-450酶系统对药物氧化过程示意图
图3-2 细胞色素P-450酶系统对药物还原过程示意图
生物转化的第二步反应是结合。多数经过氧化反应的药物再经肝微粒体的葡萄糖醛酸转移酶作用与葡萄糖醛酸结合。有些药物还能和乙酰基、甘氨酸、硫酸等结合。这些结合反应都需要供体参加,例如二磷酸尿嘧啶是葡萄糖醛酸的供体。药物在体内转化过程,举例说明见表3-1。
表3-1 药物生物转化类型举例
| 转化类型 |
转化反应通式 |
酶系 |
药物举例 |
1.氧化
脂肪族羟化
芳香族羟化
N去烷基
O去烷基
硫氧化
去硫
去卤
环氧化
醇类氧化
醛类氧化
胺类氧化
嘌呤氧化
2.还原
硝基还原
偶氮还原
醛类还原
酮类还原
3.水解
酰胺键水解
酯键水解
4.结合
葡萄糖醛酸
结合
乙酰化
|
R→ROH
Ar→ArOH
CH3
|
R1―N―R2→R1―NH―R2
R―O―CH3→ROH
O
‖
R1―S―R2→R1―S―R2
S O
‖ ‖
R1―P―R2→R1―P―R2
X OH
| |
R1―CH―R2→R1―CH―R2+HX
O
/ \
R1―CH=CH―R2→R1―CH―CH―R2
R―CH2OH→RCHO
RCHO→RCOOH
RCH2NH2→RCHO+NH2
Ar(N)→Ar(O)
ArNO2→ArNH2
Ar1―N=N―Ar2→Ar1NH2+Ar2NH2
RCHO→RCH2OH
O OH
‖ |
R1―C―R2→R1―CH―R2
R1―CONH―R2→R1COOH+R2NH2
R1COOR2→R1COOH+R2OH
载体:UDP-葡萄糖醛酸
载体:乙酰辅酶A
|
微粒体酶
微粒体酶
微粒体酶
微粒体酶
微粒体酶
微粒体酶
微粒体酶
微粒体酶
非微粒体酶
非微粒体酶
非微粒体酶
非微粒体酶
微粒体酶
微粒体酶
非微粒体酶
非微粒体酶
微粒体酶
非微粒体酶
微粒体酶
非微粒体酶
|
司可巴比妥
苯妥英
地西泮
可待因
氯丙嗪
对硫磷
氟烷
苯并芘(致癌物)
乙醇
乙醛
肾上腺素,组胺
茶碱
氯硝西泮
百浪多息
水合氯醛
纳洛酮
利多卡因,普鲁卡因胺
乙酰胆碱,普鲁卡因
氯霉素,吗啡
异烟肼
|
四、排 泄
药物在体内最后的过程是排泄(excretion),肾脏是主要排泄器官。游离的药物能通过肾小球过滤进入肾小管。随着原尿水分的回收,药物浓度上升。当超过血浆浓度时,那些极性低、脂溶性大的药物反向血浆扩散(再吸收),排泄较少也较慢。只有那些经过生物转化的极性高、水溶性代谢物不被再吸收而顺利排出。有些药物在近曲小管由载体主动转运入肾小管,排泄较快。在该处有两个主动分泌通道,一是弱酸类通道,另一是弱碱类通道,分别由两类载体转运,同类药物间可能有竞争性抑制。例如丙磺舒抑制青霉素主动分泌,使后者排泄减慢,药效延长并增强。碱化尿液使酸性药物在尿中离子化,酸化尿液使碱性药物在尿中离子化,利用离子障原理阻止药物再吸收,加速其排泄,这是药物中毒常用的解毒方法(图3-3)。
图3-3 尿液酿碱度对弱酸性(水杨酸)及弱碱性
(苯丙胺)药物在肾小管内再吸收的影响
药物可自胆汁排泄,原理与肾排泄相似,但不是药物排泄的主要途径。药物自胆排泄有酸性、碱性及中性三个主动排泄通道。有些药物在肝细胞与葡萄糖醛酸等结合后排入胆中,随胆汁到达小肠后被水解,游离药物被重吸收,称为肝肠循环(hepato-enteral circulation)。在胆道引流病人,药物的血浆半衰期将显著缩短,如氯霉素、洋地黄等。乳汁pH略低于血浆,碱性药物可以自乳汁排泄,哺乳婴儿可能受累。胃液酸度更高,某些生物碱(如吗啡等)注射给药也可向胃液扩散,洗胃是中毒治疗和诊断的措施。药物也可自唾液及汗液排泄。粪中药物多数是口服未被吸收的药物。
肺脏是某些挥发性药物的主要排泄途径,检测呼出气中的乙醇量是诊断酒后驾车的快速简便的方法。
体内药量随时间而变化的过程是药动学研究的中心问题。药量与效应的关系(量效关系)已在药效学章详述。加入时间因素就引出时量关系(time-concentration relationship)与时效关系(time-response relationship)。大多数情况下由于量效关系基本固定,在达到“平衡”后两条曲线平行一致。整体动物一次血管外给药的时量(效)曲线见图3-4。按一室模型理解,曲线升段主要是吸收过程(此时消除过程已经开始)。曲线在峰值浓度(peak concentration, Cmax)时吸收速度与消除速度相等。从给药时至峰值浓度的时间称为达峰时间(peak time, Tpeak),曲线降段主要是药物消除过程。血药浓度下降一半的时间称为消除半衰期(elimination half-life time)。血药浓度超过有效浓度(低于中毒浓度)的时间称为有效期(effective peroid)。曲线下面积(area under the curve, AUC)与吸收入体循环的药量成比例,反映进入体循环药物的相对量。AUC是血药浓度(C)随时间(t)变化的积分值:
当t1为0,t2为∞时,AUC=Co/Ke,单位是g.h.L-1。
图3-4 ,典型时量曲线图
MTC最小中毒浓度 MEC最小有效浓度
图3-5 某药剂量相等的三种制剂的生物利用度比较
F(AUC)相等,但Tpeak及 Cmax不等
生物利用度(bioavailability)是指经过肝脏首关消除过程后能被吸收进入体循环的药

剂口服后测得的量效曲线,其AUC相等(表示F值相等),但Tpeak及Cmax不等,吸收快的Cmax可能已超过最低中毒浓度,吸收慢的Cmax可能还在有效浓度以下。生物利用度是药物制剂质量的一个重要指标。
从生理学看,体液被分为血浆、细胞间液及细胞内液几个部分。为了说明药动学基本概念及规律现假定机体为一个整体,体液存在于单一空间,药物分布瞬时达到平衡(一室模型)。问题虽然被简单化,但所得理论公式不失为临床应用提供了基本规律。按此假设条件,药物在体内随时间变化可用下列基本通式表达:dC/dt=kCn。C为血药浓度,常用血浆药物浓度。k为常数,t为时间。由于C为单位血浆容积中的药量(A),故C也可用A代替:dA/dt=kCn,式中n=0时为零级动力学(zero-order kinetics),n=1时为一级动力学(first-order kinetics),药物吸收时C(或A)为正值,消除时C(或A)为负值。在临床应用中药物消除动力学公式比较常用,故以此为例如以推导和说明。
一、零级消除动力学
当n=0时,-dC/dt=KC0=K(为了和一级动力学中消除速率常数区别,用K代k),将上式积分得:
Ct=C0- Kt,C0为初始血药浓度,Ct为t时的血药浓度,以C为纵座标、t为横座标作图呈直线(图3-6),斜率为K,当Ct/C0=1/2时,即体内血浆浓度下降一半(或体内药量减少一半)时,t为药物消除半衰期(half-life time, t1/2)。
按公式1/2C0=C0-Kt1/2
∴t1/2=0.5C0/K
可见按零级动力学消除的药物血浆半衰期随C0下降而缩短,不是固定数值。零级动力学公式与酶学中的Michaelis-Menten公式相似:dS/dt=[S]Vmax/([S]+Km),式中S为酶的底物,Vmax为最大催化速度,Km为米氏常数。当[S]>>Km时,Km可略去不计,ds/dt=Vmax,即酶以其最大速度催化。零级动力学公式与此一致,说明当体内药物过多时,机体只能以最大能力将体内药物消除。消除速度与C0高低无关,因此是恒速消除。例如饮酒过量时,一般常人只能以每小时10ml乙醇恒速消除。当血药浓度下降至最大消除能力以下时,则按一级动力学消除。
图3-6 药物在体内消除过程的时量曲线
体内药物过多,超过机体最大消除能力(虚线)时为零级动力学恒速消除
体内药物降至虚线以下时为一级动力学恒比消除。插图纵坐标为对数标尺
二、一级消除动力学
当n=1时,-dC/dt=keC1=keC,式中k用ke表示消除速率常数 (elimination rate constant)。将上式积分得

可见按一级动力学消除的药物半衰期与C高低无关,是恒定值。体内药物按瞬时血药浓度(或体内药量)以恒定的百分比消除,单位时间内实际消除的药量随时间递减。消除速率常数(ke)的单位是h-1,它不表示单位时间内消除的实际药量,而是体内药物瞬时消除的百分率。例如ke=0.5h-1不是说每小时消除50%(如果t1/2=1小时则表示每小时消除50%)。按t1/2=0.693/ke计算t1/2=1.39h, 即需1.39h后才消除50%。再按 计算,1小时后体内尚存60.7%。绝大多数药物都按一级动力学消除。这些药物在体内经过t时后尚存

当n=5时,At≈3%A0,即经过5个t1/2后体内药物已基本消除干净。与此相似,如果每隔一个t1/2给药一次(A0),则体内药量(或血药浓度)逐渐累积,经过5个t1/2后,消除速度与给药速度相等,达到稳态(steady state):
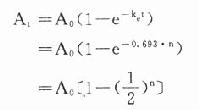
当n=5时,At≈97%A0。这一时间,即5个t1/2不因给药剂量多少而改变。具体数值见表3-2。
表3-2 一级动力学药物在体内消除量及累积量
药物自体内消除的一个重要指标是血浆清除率(plasma clearance,Cl), 是肝肾等的药物消除率的总和,即单位时间内多少容积血浆中的药物被消除干净,单位用L·h-1(也有人用 ml·min-1,和肌酐消除率一致)或按体重计算 L·kg-1·h-1 。按定义,CL=RE/Cp,RE是消除速率(rate of elimination),即单位时间内被机体消除的药量,Cp为当时的血浆药物浓度。由于RE非固定值也不易检测,故常用表观分布容积 (apparent volume of distribution, Vd)计算。 Vd是指静脉注射一定量(A)药物待分布平衡后,按测得的血浆浓度计算该药应占有的血浆容积。事实上静注药物后未待分布平衡已有部分药物自尿排泄及(或)在肝转化而消除,故必需多次检测Cp,作时量曲线图,将稳定下降的消除段向O时延升至和Y轴交点以求得理论上静注药量A在体内分布平衡时的血浆浓度C0,以此算出Vd=A/C0(图3-7)。按RE=keA,Cp=A/Vd,故Cl=keVd。在一级动力学的药物中,Vd及Cl是两个独立的药动学指标,各有其固定的数值,互不影响,也不因剂量大小而改变其数值。Vd是表观数值,不是实际的体液间隔大小。除少数不能透出血管的大分子药物外,多数药物的Vd值均大于血浆容积。与组织亲和力大的脂溶性药物其 Vd可能比实际体重的容积还大。Cl也不是药物的实际排泄量。它反映肝和(或)肾功能,在肝和(肾)功能不足时Cl值会下降,因为Cl是肝肾等消除能力的总和。肝清除率虽然难测,但有重要的理论意义。肝清除率小的药物,首关消除少,其口服生物利用度大,但易受肝功能,血浆蛋白结合力及肝药酶诱导或抑制药的影响。肝清除率大的药物,首关消除多,其口服生物利用度小。有些药物的肝清除率很高,接近肝血流量,称为灌流限制性清除,其肝清除率受肝血流量影响较大。药物以原形自肾消除的百分率比较容易测定。自肾排泄多的药物易受肾功能影响,自肾排泄少的药物易受肝功能影响。医生可以据此在肝或肾功能不足病人适当调整剂量。在零级动力学的药物中,RE以恒速消除,不随Cp下降而改变,故Cl 值不固定,与Cp成反比。
图3-7 表观分布容积计算法
C
0是静注药量A在0时理论上的血药浓度
Cl值实际上常用静脉或肌肉注射药物A后测定Cp,绘出时量曲线,算出AUC再按CL=A/AUC取得。因为AUC=C0/ke,代入得
CL=keVd=C0Vd/AUC=A/AUC。
三、连续恒速给药
临床治疗常需连续给药以维持有效血药浓度。在一级动力学药物中,开始恒速给药时药物吸收快于药物消除,体内药物蓄积。按 计算约需5个t1/2达到血药稳态浓度(Css)(图3-8),此时给药速度(RA)与消除速度(RE)相等。
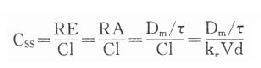
(τ为给药间隔时间)可见Css随给药速度(RA=Dm/τ)快慢而升降,到达Css时间不因给药速度加快而提前,它取决于药物的ke或t1/2。据此,可以用药物的keVd或Cl计算给药速度以达到所需的有效药物浓度。静脉恒速滴注时血药浓度可以平稳地到达Css。分次给药虽然平均血药浓度上升与静脉滴注相同,但实际上血药浓度上下波动(图3-8)。分药间隔时间越长波动越大,其峰值浓度
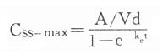 ,谷值浓度 Css-min=Css-maxe 。如果实际Css过高或过低,可以按已达到的Css与需要达到的Css比值调整给药速度,即Css(已达到的)/Css(需要的)=RA(现用的)/RA(将调整的)
,谷值浓度 Css-min=Css-maxe 。如果实际Css过高或过低,可以按已达到的Css与需要达到的Css比值调整给药速度,即Css(已达到的)/Css(需要的)=RA(现用的)/RA(将调整的)
图3-8 连续恒速给药时的时量曲线
约经5个半衰期血药浓度达到稳态。给药间隔越短,
血药浓度波动越小。给药剂量越大,血药浓度越高
A.静脉滴注,Dm/t1/2 B.肌肉注射,Dm/t1/2
C.肌肉注射,1/2Dm/2t1/2。Dm维持剂量
但从调整剂量时开始需再经过5个t1/2方能达到需要的Css。
在病情危重需要立即达到有效血药浓度时,可于开始给药时采用负荷剂量(loading dose,D1),因为
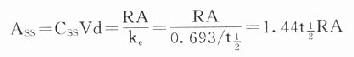
Ass就是负荷剂量。可将第一个t1/2内静脉滴注量的1.44倍在静脉滴注开始时推注入静脉即可立即达到并维持Css。在分次恒速给药达到Css时,体内Ass是维持剂量(maintenance dose, Dm)与体内上一剂量残留药物的和,即

当给药间隔时间τ=t1/2时,

即每隔一个t1/2给药一次时采用首剂加倍剂量的D1可使血药浓度迅速达到Css。
理想的给药方案应该是使Css-max略小于最小中毒血浆浓度(MTC)而Css-min略大于最小有效血浆浓度(MEC),即血药浓度波动于MTC与MEC之间治疗窗,这一Dm可按下列公式计算:
Dm=(MTC - MEC)Vd
负荷剂量计算法与上同,即D1=ASS=1.44t1/2 RA=1.44t1/2 Dm/τ,τ为给药间隔时间。τ可按一级消除动力学公式 推算得
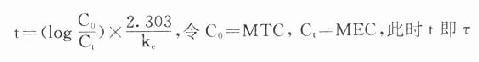
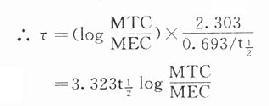
因此可以根据药物的MTC及MEC利用这些公式计算出D1,Dm及τ。注意此时 τ≠t1/2,D1≠2Dm(图3-9)。
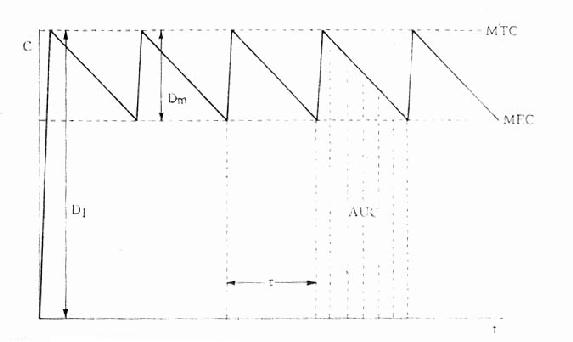
图3-9 负荷剂量、维持剂量、给药间隔与血药浓度关系
Dm维持剂量所形成的C D1负荷剂量所形成的C
在零级动力学药物中,体内药量超过机体最大消除能力。如果连续恒速给药,RA>RE,体内药量蓄积,血药浓度将无限增高。停药后消除时间也较长,超过5个t1/2。因为t1/2=0.5C0/K,达到C0越高t1/2越长。
临床用药可根据药动学参数如Vd、Cl、ke、t1/2及AUC等按以上各公式计算剂量及设计给药方案以达到并维持有效血药浓度。除了少数t1/2特长或特短的药物,或零级动力学药物外,一般可采用每一个半衰期给于半个有效量(half dose at half life interval)并将首次剂量加倍是有效、安全、快速的给药方法。
有些药在体内转化为活性产物则需注意此活性产物的药动学,如果活性产物的消除是药物消除的限速步骤的话,则应按该产物的药动学参数计算剂量及设计给药方案。
四、一级药动学指标间的相互关系
1.F=A/D×100%口服剂量(D)由于不能100%吸收及存在首关消除效应,能进入体循环的药量(A)只占D的一部分,这就是生物利用度(F)。药动学计算时应采用绝对生物利用度,相对生物利用度作为评比药物制剂质量的指标。生物利用度还包括吸收速度问题,达峰时间(Tpeak)是一个参考指标。
2.A=C·Vd或C=A/Vd体内药量(A)与血药浓度(C)比值固定,在许多药动学公式中,A与C可以通用,如At=也可用Ct=。
3.Cp=[D]+[DP] 血浆中药物有游离型(D)与血浆蛋白结合型(DP),定量测定时需将血浆蛋白沉淀除去,故通常所说的血浆药物浓度(Cp)是指[D]与[DP]的总和。只有透析法或超离心法才可能将二者分离以计算药物的血浆蛋白结合率[DP]/([D]+[DP])×100%。
4.
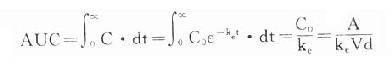 曲线下面积(AUC)是一个可用实验方法测定的药动学指标。它反映进入体循环药量的多少。时量曲线某一时间区段下的AUC反映该时间内的体内药量。AUC是独立于房室模型的药动学参数,常用于估算血浆清除率(Cl)。
曲线下面积(AUC)是一个可用实验方法测定的药动学指标。它反映进入体循环药量的多少。时量曲线某一时间区段下的AUC反映该时间内的体内药量。AUC是独立于房室模型的药动学参数,常用于估算血浆清除率(Cl)。
5.ke=0.693/t1/2=RE/A=CL/Vd 消除速率常数是药物瞬时消除的百分率而不是单位时间药物消除速率(RE),是决定t1/2的参数,但其本身又取决于Cl及Vd,故不是独立的药动学指标。
6.Vd=A/C0=A/AUC ke 表现分布容积(Vd)是独立的药动学指标,不是实际的体液容积,取决于药物在体液的分布。Vd大的药物与组织蛋白结合多,主要分布于细胞内液及组织间液。Vd小的药物与血浆蛋白结合多,较集中于血浆。Vd不因A多少而变化。
7.CL=keVd=RE/Cp=A/AUC 血浆清除率(Cl)是肝肾等清除率的总和,也不是实际的药物消除速率(RE),是另一个独立于A的重要药动学指标,但受肝肾功能的影响。
8.t1/2=0.693/ke=0.693Vd/CL 血浆药物消除半衰期(t1/2)是一个非常实用的药动学指标,虽然独立于A,但受Cl及Vd双重制约,Cl大时t1/2短,Vd大时t1/2长。例如庆大霉素Cl小(60ml·min-1), Vd也小(0.25L·kg-1),其t1/2不长(2~3h)。氯喹Cl大(700ml·min-1),Vd也大(185L·kg-1),其t1/2并不短(8天)。药物在吸收及分布过程中也有半衰期,分别用t1/2a及t1/2α表示。
9.稳态时RA=RE=CSS·Cl=CSS·Vd·ke
故
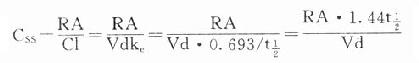 CSS是恒速连续给药达到稳态时平均血药浓度,应该和预期的有效浓度相等。必要时可以按达到的CSS与预期的CSS比值调整剂量或给药速度(RA)。
CSS是恒速连续给药达到稳态时平均血药浓度,应该和预期的有效浓度相等。必要时可以按达到的CSS与预期的CSS比值调整剂量或给药速度(RA)。
10.
 分次定时定量给药时,CSS上下波动。当每t1/2给药一次时,其峰值(Cssmax)与谷值(Css-min)的比值为2,缩短给药间隔可以减少CSS波动。
分次定时定量给药时,CSS上下波动。当每t1/2给药一次时,其峰值(Cssmax)与谷值(Css-min)的比值为2,缩短给药间隔可以减少CSS波动。
11.
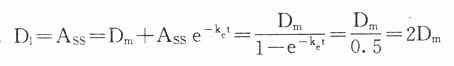 每t1/2给药一次时,首次给予加倍剂量,即负荷剂量(D1)可以立即达到CSS。
每t1/2给药一次时,首次给予加倍剂量,即负荷剂量(D1)可以立即达到CSS。
五、房室模型
以上所述各种药动学公式都是将机体视为一个整体空间,假设药物在其中转运迅速,瞬时达到分布平衡的条件下推导而得的。实际上机体绝非如此简单,不仅有血浆、细胞外液及细胞内液等间隔,而且各组织细胞间存在着无数的区间。静脉注射药物的时量(对数标尺)关系并非直线,而是一条由无数区段组成的连续弧线。粗略地看可见早期一段快速下降,后来才逐渐稳定缓慢下降。这是因为药物进入血液循环后快速向组织分布,首先进入血注量大的肺、肾、心、脑等器官,然后再向其他组织分布,最后达到平衡(假平衡)。因此设想机体由几个互相连通的房室(compartment)组成。这个房室不是解剖学上分隔体液的房室,而是按药物分布速度以数学方法划分的药动学概念。多数药物按二房室模型转运(少数单房室或多房室),中央室大致包括血浆及那些血流量多的器官,周边室包括机体其余部分,界限并不明确。时量曲线因此也只能大致分为分布相及消除相两个指数衰减区段(图3-10)。其药动学规律与单房室不同,如C=Ae-αt +Be-βt,α及β分别为分布相(A)及消除相(B)的消除速率常数。而且在分布相中Vd逐渐增大,ke(α)逐渐减少,t1/2逐渐延长,因此药动学计算需要特殊处理。即使在消除相,血药浓度稳定线性下降,各组织浓度及其下降速度也不尽相等,故称假平衡。可见问题非常复杂。
图3-10 二房室模型时量曲线
A.分布相(实线)及分布曲线(虚线)
B.消除相(实线)与消除曲线(虚线)
正由于问题过于复杂,临床应用诸多不便,实际运算也存在诸多困难。房室模型并非药物固有的药动学指标,机体也无此解剖学间隔,即使运用电子计算机拟合也不一定获得明确的划分。用同一药物试验,在某些人呈二室模型,另些人可能呈一室或三室模型。同一药物静脉注射时呈二室模型而口服则呈单一房室模型。在分布相时药物实际上已开始消除,到达消除相时可能已有相当分量的药物已被消除。如果用血管外给药(口服、肌注等)分布相常被吸收相掩盖。这些时相的划分仅靠血药浓度的测定。如果早期(此时血药浓度变化较快)取样间隔过疏,很难据此准确划分时相,因此,越来越多的临床家及研究者逐渐放弃房室模型而转向采用适用于所有药物的无房室方法(noncompartmental medtod)来解决实际问题,对此,有待今后深入学习。
从另一方面看,时量曲线在达到假平衡后已呈单一指数衰减的直线,此时房室划分已无需要,可以按β值计算t1/2及其他实用的药动学指标。
AUC是与房室无关的药动学指标,可用实验方法测定。AUC(0-∞)=C0/ke,或AUC(T-∞)=Ct/β,T是消除相开始的时间。再用AUC算出Vd及Cl:
Vd=A/AUCβ,CL=A/AUC

 中医世家
中医世家 浦 标 网
浦 标 网 河南大学精品课程
河南大学精品课程 图书资料室
图书资料室