弥散性血管内凝血(disseminatedor diffuse intravascular coagulation, DIC)是指在某些致病因子作用下凝血因子或血小板被激活,大量可溶性促凝物质(soluble thromboplastin)入血,从而引起一个以凝血功能失常为主要特征的病理过程(或病理综合征)。此时微循环中有纤维蛋白性微血栓或血小板团块形成,同时一系列血浆凝血因子被消耗,血小板减少,并有继发性纤维蛋白溶解(纤溶)过程加强。在临床上,DIC患者主要表现为出血、休克、脏器功能障碍和贫血。
由于DIC的发病机制和临床表现比较复杂,因此长期以来,曾经有过许多不同的命名。近年来,某些学者认为,为了更全面地表达此病理过程的变化特点,建议将DIC称为消耗性血栓-出血性疾病(cosumptive thrombohemorrhagicdisorders)。本章继续使用弥散性血管内凝血的命名。
第一节 弥散性血管内凝血的原因和发病机制
正常机体的血液呈液体状态,在心、血管内流动不止,同时也不发生血凝。这是由于机体存在着凝血、抗凝血和纤维蛋白溶解系统,它们处于动态平衡状态。其中以凝血过程和纤维蛋白溶解过程最为重要,二者保持着极为密切的关系。
DIC的病因众多,引起DIC的发病机制为复杂,但其中以血管内皮细胞的损伤与组织损伤为最重要。
一、血管内皮细胞损伤、激活凝血因子Ⅻ,启动内源性凝血系统
细菌、病毒、螺旋体、高热、抗原抗体复合物、休克时持续的缺血、缺氧和酸中毒、败血症时的细菌内毒素等,在一定条件下皆可使血管内皮细胞发生损伤,使其下面的胶原暴露。胶原、内毒素等均为表面带负电荷的物质,当无活性的凝血因子Ⅻ与这些物质表面发生接触后,其精氨酸残基上的胍基在负电荷影响下分子构型发生改变,它的活性部分——丝氨酸残基暴露,所以因子Ⅻ被激活(此种激活方式称接触激活或固相激活)。另外,也可能在激肽释放酶、纤溶酶或胰蛋白酶等可溶性蛋白水解酶的作用下,因子Ⅻ或Ⅻa通过酶性水解(酶性激活或液相激活)而生成Ⅻf。胶原等激活因子Ⅻ的过程开始时进行得较为缓慢,但因Ⅻ的碎片(Ⅻf),即激肽释放酶原激活物(predallidreinactivator, PKA)可把血浆激肽释放酶原(prekallikrein)激活成激肽释放酶(kallikrein),后者又能反过来使因子Ⅻ进一步活化,从而使内源性凝血系统的反应加速(图9-1)。Ⅻa和Ⅻf还可相继激活纤溶、激肽和补体系统,从而进一步促进DIC发展。
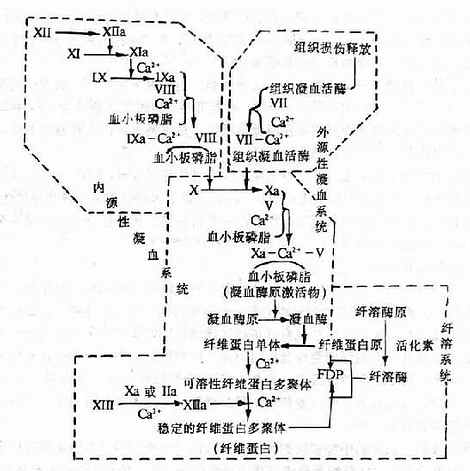
图9-1 血液凝固过程及纤溶系统
以内毒素血症为例,此时,除内毒素可直接激活因子Ⅻ外,内毒素还可引起血管内皮细胞损伤,基底膜暴露后,胶原、胶原与某些糖蛋白的复合物或另一些结缔组织成分也可激活因子Ⅻ。此外,还有某些酸糖脂(acidic glycolipids),硫酸脂(sulfatides),氨基葡聚糖(glycosaminoglycans)或另外一些特殊的因子Ⅻ激活物也可自损伤的内皮细胞释放,因此内源性凝血系统启动。在家兔内毒素引起的DIC中常可有内皮细胞脱落,在循环血中有内皮细胞出现。抗原抗体复合物粘附在肾小球等微血管壁上时,可引起血管内皮细胞损伤。血管炎时也可继发血管内皮细胞损伤,进一步触发DIC。
此外,在内皮细胞受损时,血小板与内皮下结缔组织中的胶原接触后可以产生胶原诱导的促凝活性,此时,因子Ⅺ可不通过Ⅻa而直接被激活,从而推动凝血连锁反应,引起DIC。
表9-1不同的人体组织中凝血因子Ⅲ的含量
| 组织 |
含量 (μ/mg) |
| 肝 |
10 |
| 肌肉 |
20 |
| 脑 |
50 |
| 肺 |
50 |
| 胎盘 |
2,000 |
| 蜕膜 |
2,000 |
二、组织严重破坏使大量组织因子进入血液,启动外源性凝血系统
在外科大手术、严重创伤、产科意外(如胎盘早期剥离、宫内死胎等)、恶性肿瘤或实质性脏器的坏死等情况下均有严重的组织损伤或坏死,所以大量促凝物质入血(表9-1),其中尤以组织凝血活酶(tissuethromboplastin,即凝血因子Ⅲ,或称组织因子)较多。这些促凝物质可通过外源性凝血系统的启动引起凝血。
组织因子是一种脂蛋白复合物,含有大量磷脂。当它进入血浆后。血浆中的钙离子将因子Ⅶ连接于组织因子的磷脂上,形成复合物,后者可使凝血因子X活化为Xa,并与Ca2+、因子V和血小板磷脂相互作用而形成凝血酶原激活物,然后通过与内源性凝血系统后阶段相同的途径,完成凝血的化学反应(图9-1)。
以宫内死胎为例,当胎儿的坏死组织在子宫内滞留超过5周,DIC的发生率可达50%左右,这是因为坏死的胎儿组织释放组织因子,后者大量进入母体循环,启动外源性凝血系统。此外有人证明,当肿瘤组织坏死时,释放出一种蛋白酶,如某些腺癌能分泌一种含有唾液酸的粘蛋白,它可直接激活X因子,从而启动凝血连锁反应。
三、血细胞大量破坏
红细胞大量破坏时常可发生DIC。急性溶血,如大量(>50ml)误型输血、药物引起的免疫性溶血时,抗原-抗体复合物的形成对凝血起主要作用。因为据报道,在蚕豆病中由非免疫因素引起的血管内溶血以及实验性血红蛋白尿等情况下常常不产生DIC。因此,一般认为只有在红细胞大量破坏伴有较强的免疫反应时,DIC才比较容易发生。此外,红细胞大量破坏释出的ADP与DIC的发生有关,因为后者触动了血小板释放反应,使大量血小板第3因子(PF3)入血,促进凝血过程。红细胞膜内大量的磷脂既有直接的促凝作用,又能促进血小板的释放而间接促进凝血过程。
实验研究证明,正常的中性粒细胞和单核细胞内有促凝物质。在内毒素或败血症所引起的DIC时内毒素可使中性粒细胞合成并释放组织因子,同时有大量白细胞在肺血管中停滞,并释放出大量促凝物质(可能就是组织因子),这些物质进入体循环进一步加速了凝血反应,所以肺似乎起了凝血的放大作用。大量促凝物质从崩解的白细胞中释放出来,从肺血管经左心进入主动脉后,肾脏首先受累,因此肾脏微血栓发生率较高,病变程度较重。另外,在病人患急性早幼粒细胞性白血病时,此类白血病细胞浆中含有凝血活酶样物质,当白血病细胞大量坏死或经化疗杀伤时,这些物质就大量释放入血,通过外源性凝血系统的启动而引起DIC。
血小板在DIC的发生发展中起着重要的作用。内毒素、免疫复合物、颗粒物质、凝血酶等都可直接损伤血小板,促进它的聚集。微血管内皮细胞的损伤,内皮下胶原和微纤维的暴露是引起局部血小板粘附、聚集、释放反应的主要原因,这是因为是构成胶原的肽链中,存在着一个与血小板粘附有关的活性部位。血小板表面的糖蛋白Ib(glycoprotein Ib, GPIb)对血小板粘附起重要作用,GPIb通过血浆因子(如Ⅷ相关抗原/vonWillebrand因子,Ⅷ/VWF因子)使血小板与内皮下组织粘连。另外,由于血小板膜上的另一些糖蛋白(GPⅡb,GPⅡa)能结合于纤维蛋白原,后者通过与钙离子的连接,在血小板之间“搭桥”,使血小板聚集。血小板发生粘附、释放和聚集后,除有血小板微集物形成(microaggregateformation,图9-2)堵塞微血管外,还能进一步激活血小板的凝血活性,促进DIC的形成。但是在不同病因所引起的DIC中,血小板所发挥的作用并不一致,它可以起原发的作用,如血栓性血小板减少性紫癜,在发病开始时即可由免疫反应等原因使血小板发生聚集,其中PF3(血小板第3因子)能加速凝血酶原的激活,PF4(血小板第4因子)能中和肝素并使可溶性纤维蛋白多聚体沉淀。β-血栓球蛋白也具有促凝作用,从而加速血液凝固,形成微血栓。但是,一般来说,在DIC发病中,血小板多起继发的作用。在外源性凝血系统被激活所致的DIC中,血小板不起主要作用,在内毒素引起的DIC中,血小板对白细胞的促凝机制还有促进作用。实验证明,人类白细胞与内毒素同时孵育后所产的促凝活性可因加入血小板而增强,这可能是血小板膜上的脂蛋白、白细胞及某些凝血因子相互作用造成的。
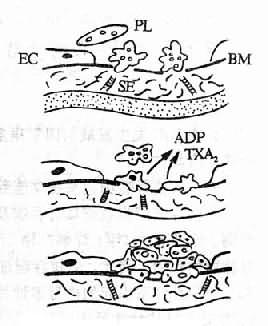
图9-2 血小板微聚物形成机制示意图
PL血小板 EC 内皮细胞SE 内皮下组织 BM 基底膜
四、其它促凝物质进入血液
一定量的羊水、转移的癌细胞或其它异物颗粒进入血液可以通过表面接触使因子Ⅻ活化,从而激活内源性凝血系统。急性胰腺炎时,蛋白酶进入血液能促使凝血酶原变成凝血酶。毒蛇咬伤时,某些蛇毒如蝰蛇的蛇毒含有一种蛋白酶,它可直接水解凝血酶原形成凝血酶。响尾蛇的蛇毒可直接使纤维蛋白原凝固。抗原抗体反应也可以引起DIC,这可能是抗原抗体复合物能激活因子Ⅻ或损伤血小板引起血小板聚集并释放促凝物质(如血小板因子等)所致。补体的激活在DIC的发生发展中也起着重要的作用。有人发现,给正常动物静脉注射内毒素后,出现动脉血压下降,血小板及纤维蛋白原等凝血因子减少;但如事先耗竭动物的补体,然后再注射内毒素,则该动物血压改变不明显,DIC实验室检查的异常变化轻微,存活率比未去除补体的动物高,由此可见补体系统在内毒素引起的的DIC中也起一定的作用。补体系统激活的产物C3a、C5a可引起组织肥大细胞、血液嗜碱性粒细胞的脱颗粒反应,从而释放5-羟色胺、组胺等物质。组胺能使毛细血管、微静脉等部位的血管内皮细胞收缩,内皮细胞之间的裂隙扩大,内皮下的胶原暴露,促使内源性凝血系统激活。此外,补体系统激活后C3b还可通过人单核细胞上的C3b受体而使凝血因子Ⅲ的释放增多。补体系统还能直接或间接地促进血小板释放PF3。
上述各种所致DIC的机制如图9-3所示。它们常常综合或相继起作用。
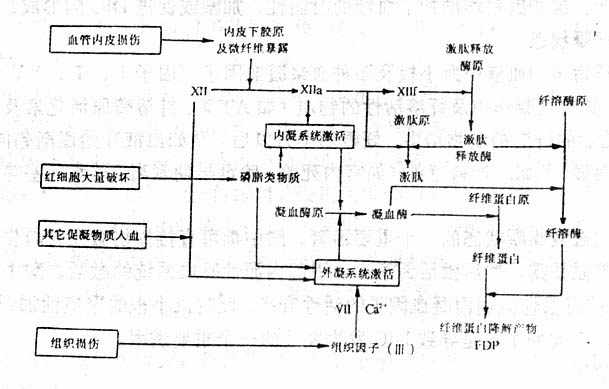
图9-3 DIC的发病机制
第二节 影响弥散性血管内凝血
发生发展的因素
影响DIC发生发展的因素很多,应该引起警惕,尽可能及早采取相应的措施以防止、减轻或排除其作用。
一、单核吞噬细胞系统功能受损
单核吞噬细胞系统具有吞噬及清除循环血液中的凝血酶、其它促凝物质、纤维蛋白、纤维溶酶、纤维蛋白(原)降解产物(fibrin or fibrinogen degradation product, FDP)以及内毒素等物质作用。因此,单核吞噬细胞系统的严重功能障碍会促使DIC的形成。例如在严重的革兰氏阴性细菌引起的内毒素性体克中,单核吞噬细胞系统可因吞噬大量坏死组织、细菌或内毒素而使其功能处于“封闭”状态;同样,在严重的酮症酸中毒时,大量脂质有时也可“封闭”单核吞噬细胞系统,这时机体再与内毒素接触就易于发生DIC。
全身性Shwartzman反应(generalized Shwartzman reaction,GSR)是给家兔间隔24小时静脉内各注射一次小剂量内毒素,在接受第二次注射后家兔就发生休克、出血倾向,甚至因急性肾功能衰竭而死亡。死亡解剖发现各重要脏器的微循环中常有纤维蛋白性微血栓,而且由此产生相应组织的缺血坏死,其中尤以肾、肺、肝等最为明显。如果第一次注射时用具有封闭单核吞噬细胞系统作用的二氧化钍代替内毒素,则第二次注射小剂量内毒素后同样发生DIC。目前一般认为GSR的发生机制之一是由于第一次内毒素注射后单核吞噬细胞系统吞噬了内毒素和纤维蛋白而被“封闭”,因此第二次注射时,单核吞噬细胞系统清除激活的凝血因子的能力降低,并无法使内毒素灭活。内毒素具有损伤血管内皮、激活血小板及凝血因子Ⅻ、促使血小板聚集和收缩血管等作用,故能引起DIC。
二、肝功能严重障碍
肝功能严重障碍时肝脏产生的某些抗凝物质如抗凝血酶Ⅲ(antithrombin Ⅲ,AT-Ⅲ)减少,引起肝功能障碍的某些病因,如肝炎病毒、某些药物、抗原抗体复合物等均可激活凝血因子。肝细胞如有大量坏死,又可释放组织凝血活酶(因子Ⅲ)样物质。此时机体经肝脏处理乳酸的能力降低。这些因素均增加了血液的凝固性,加剧或促进DIC的形成。
三、血液的高凝状态
妊娠后三周开始孕妇血液中血小板及多种血浆凝血因子(因子Ⅰ、Ⅱ、Ⅴ、Ⅷ、Ⅸ、Ⅹ及Ⅻ等)增多,而具有抗凝作用及纤溶活性的物质(如ATⅢ、纤溶酶原活化素及尿中尿激酶等)降低,来自胎盘的纤溶抑制物增多。妊娠四个月以后,孕妇血液开始逐渐趋向高凝状态,到妊娠末期最为明显。因此,产科意外(如宫内死胎、胎盘早期剥离、羊水不栓塞等)时DIC的发生率较高。
酸中毒是引起血液高凝状态的一个重要因素。酸中毒可直接损伤微管内皮细胞,使内皮下的微纤维与胶原暴露,然后激活因子Ⅻ,引起内原凝血系统的激活。酸中毒时,血液pH降低,肝素的抗凝活性减弱而凝血因子的活性升高,此时血小板的聚集性加强,由它释放的促凝因子增加,因此酸中毒是导致DIC发生发展的一个重要诱因。
四、微循环障碍
休克导致的严重微循环障碍,常有血流淤滞,血细胞聚集,血液甚至可呈淤泥状(sludging)。巨大血管瘤时对毛细血管中血流极度缓慢,血流出现涡流,再加上局部内皮细胞损伤与酸中毒,这些因素均有利于DIC的发生。低血容量时,由于肝、肾等脏器处于低灌流状态,无法及时清除某些凝血或纤溶产物,这也是促成DIC发生的因素。
五、其它
不恰当地应用纤溶抑制剂如6-氨基已酸(ε-aminocaproic acid, EACA)、对羧基苄胺(paminomethyl benzoic acid,PAMBA)等药物造成纤溶系统的过度抑制、血液粘度增高时也会促进DIC形成。DIC的发生可能还与病人当时的微血管功能状态有关,例如,有实验证明大剂量长时间地使用α受体兴奋剂会促使DIC形成,但是对其发生机制还未完全阐明。
此外,DIC的发生发展还与促凝物质进入血液的数量、速度和途径有关。促凝物质进入血液少而慢时,如机体代偿功能(如吞噬功能等)健全,可不发生或仅表现为症状不明显的慢性型DIC;促凝物质入血过多过快,超过机体代偿能力时,则可引起急性DIC。此外,DIC的定位与促凝物质入血的途经有重要关系。动物实验证明,股静脉内注入凝血酶所引起的DIC,微血栓的分布以肺为主,主动脉内注入则微血栓主要在肾。
第三节 弥散性血管内凝血的发展
过程(分期)及分型
一、分期
DIC是一个病理过程,根据它的病理生理特点及发展过程,典型者一般可经过三期:
(一)高凝期
由于凝血系统被激活,所以多数患者血中凝血酶含量增多,导致微血栓的形成,此时的表现以血液高凝状态为主。
(二)消耗性低凝期
由于凝血系统被激活和微血栓的形成,凝血因子、血小板因消耗而减少,此时常伴有继发纤溶。所以有出血的表现。
(三)继发性纤溶亢进期
在凝血酶及Ⅻa的作用下,纤溶酶原活化素被激活,从而使大量纤溶酶原变成纤溶酶;此时又有FDP的形成,它们均有很强的纤溶和/或抗凝作用,所以此期出血十分明显。
二、分型
由于引起DIC的原因很多,其发生发展速度也不相同,因此又可将DIC分为以下各型:
(一)按DIC发生快慢分为急性型、亚急性型与慢性型
主要和致病因素的作用方式、强度与持续时间长短有关。当病因作用迅速而强烈时,DIC表现为急性型;相反,作用缓慢而持续时,表现为慢性型或亚急性型。各型的主要特点如下:
1.急性型 DIC可在几小时或1~2天内发生,常见于各种严重的感染,特别是革兰氏阴性菌感染引起的败血症性休克、血型不合的输血、严重创作、移植后急性排异反应等。此时,临床表现明显,常以休克和出血为主,患者的病情迅速恶化,分期不明显,实验室检查结果明显异常。
2.亚急性型 DIC在数天内逐渐形成,常见于恶性肿瘤转移、宫内死胎等患者,表现介于急性型和慢性型之间。
3.慢性型常见于恶性肿瘤、胶原病、慢性溶血性贫血等疾病。此时,由于机体有一定的代偿能力,单核吞噬细胞系统的功能也较健全,所以各种异常表现均轻微而不明显。病程较长,临床诊断较困难,常常以某脏器功能不全的表现为主,有时仅有实验室检查异常,所以出现亚临床型的(subclinical)表现,此类DIC往往在尸解后作组织病理学检查时才被发现。在一定条件下,可转化为急性型。
(二)按DIC代偿情况分为代偿型、失代偿型和过度代偿型
在DIC发生发展过程中,血浆凝血因子与血小板不断消耗,但是骨髓和肝可通过增加血小板和血浆凝血因子的生成而起代偿作用。此时肝脏生成纤维蛋白原的能力可增加5倍,骨髓生成血小板的功能可增国10倍,因此根据凝血物质的消耗与代偿性生成增多之间的对经关系。可将DIC分为以下三型:
1.代偿型凝血因子与血小板的消耗与生成间基本上保持平衡状态。主要见于轻度DIC。此型患者可无明显临床表现或仅有轻度出血和血栓形成的症状。实验室检查无明显异常(如纤维蛋白原无明显减少),易被忽视。但如病情持续加重,则可转化为失代偿型。
2.失代偿型凝血因子和血小板的消耗超过生成。主要见于急性DIC。此型患者出血、休克等表现明显,实验室检查发现血小板和纤维蛋白原等凝血因子均明显减少。
3.过度代偿型机体代偿功能较好,凝血因子和血小板的生成迅速,甚至超过消耗。因此有时出现纤维蛋白原等凝血因子暂时升高的表现。主要见于慢性DIC或DIC恢复期。此型患者出血或栓塞症状可不太明显,但与代偿型相似,在致病因子的性质和强度发生改变时,也可转化为典型的失代偿型。
至于局部型的DIC,主要是指局限于某一脏器的多发性微血栓症,但全身仍有轻度的血管内凝血存在,多见于静脉瘤、主动脉瘤,心脏室壁瘤、人造血管、体外循环、器官移植后的排异反应等,此时常在病变局部有凝血过程的激活。因此严格地说,这是全身性DIC的一种局部表现。
第四节 弥散性血管内凝血时的机能代谢变化与临床表现
DIC时,各种典型病理变化及临床表现主要发生在急性、严重的DIC。形成这些变化的主要基础是凝血酶的生成增加、某些凝血因子的激活、消耗,纤维蛋白性微血栓的形成,以及继发性纤溶的增强。因此其病理与临床表现复杂多样,并随原发疾病的不同而异,但是在各种表现中尤以出血及微血管中微血栓的形成最为突出。
一、出血
虽然DIC病人典型的病理变化是微血栓形成,但是病人最初的临床现表现为出血,引起出血的机制有以下可能:
(一)凝血物质的消耗
在DIC发生发展过程中,各种凝血因子和血小板大量消耗,特别是纤维蛋白原、凝血酶原、因子Ⅴ、Ⅷ、Ⅹ、Ⅷ的血小板普遍减少。因此曾有人将DIC称为消耗性凝血病(consrmptive coagulopathy)。此时。因凝血物质大量减少,因而凝血过程受阻。
(二)纤溶系统的激活
DIC时在凝血系统激活后,常有继发性纤溶系统的激活。这主要是由于在凝血过程中,通过酶性激活(蛋白酶作用造成酶性水解)由Ⅻa形成Ⅻf,Ⅻf使激肽释放酶原转变成激肽释放酶,后者使纤溶酶原变为纤溶酶。一些富含纤溶酶原激活物的器官(如子宫、前列腺、肺等)因血管内凝血而发生变性坏死时,激活物便大量释放入血而激活纤溶系统。血管内皮细胞受损、缺氧、应激等也皆可激活纤溶系统,导致纤溶酶增多。纤溶酶除能使纤维蛋白(原)降解外,还能水解凝血因子Ⅴ、Ⅷ和凝血酶原等,故这些凝血因子进一步减少,从而引起凝血障碍和出血。
(三)纤维蛋白(原)降解产物的形成
凝血过程的激活以及继发性纤溶过程的启动使血中纤溶酶增多,纤维蛋白(原)被降解。纤维蛋白原在纤溶酶作用下先从其分子的Bβ链上裂解出一个小肽,然后又在Aα链上裂解出碎片A、B、C和H,留下的片段即X(分子量240~260KD),后者再在纤溶酶作用下不断裂解先后产生Y(分子量150KD)、D(分子量100KD)及E(分子量50KD)片段。它们统称为纤维蛋白原降解产物(FgDP)。纤维蛋白在纤溶酶作用下形成X'、Y’、D、E’片段,各种二聚体、多聚体及复合物,统称其为纤维蛋白降解产物(FDP)。两类FDP的功能特性基本相似,其中X,Y碎片可与纤维蛋白单体聚合,从而抑制纤维蛋白多聚体生成;Y、E碎片有抗凝血酶作用;D碎片抑制纤维蛋白单体聚合;大部分FDP均抑制血小板的粘附和聚集,因此FDP可通过强烈的抗凝作用引起出血。
临床上一般常用血浆鱼精蛋白副凝试验(plasma protamineparacoagulation test,3P试验)检查FDP存在,其主要原理为纤维蛋白原在凝血酶作用下形成许多纤维蛋白单体,后者在凝因血因子Ⅻ(纤维蛋白稳定因子)作用下形成纤维蛋白。纤维蛋白在纤溶酶作用下分解为X'、Y'、D、E'碎片,这些碎片(主要是X’碎片)可与纤维蛋白单体形成可溶性纤维蛋白单体复合物(soluble fibrinmonomer complex, SFMC)病人血浆中如有SFMC存在,则在体外加入鱼精蛋白后,此种现象称副凝现象。DIC病人血浆中由于有SFMC的存在,3P试验常呈阳性,所以此试验主要是反映SFMC和纤维蛋白降解产物中X'片段的试验。晚期DIC病人血浆中X'片段减少,D、E'明显增多,因此3P试验反而呈阴性。
临床上DIC病人可有轻重不等的多部位出血倾向(图9-4)。病理形成上既可有血管内凝血,也可有出血的表现。实验室检查有凝血时间和凝血酶原时间延长,纤维蛋白原和血小板减少等发现。出血发生在皮肤时,常可见到出血斑或局部坏死,它与周围皮肤分界清楚,边缘不规则,这种现象反映了皮肤下阻塞的终末微动脉的分布,如果较大的血管发生阻塞,则这些病变可发展形成出血性大疱或融合成片,但治疗及时、恰当,也可吸收。在重症病例,出血特别严重时,可以表现为手指或脚趾的坏疽,有时可出现对称性坏死性病变。
出血也可在静脉穿剌部位。血尿常见。此外也可出现牙龈和鼻出血。出血严重而剧烈时可引起死亡,而且用一般止血药物治疗无效。出血常可成为DIC的最初或主要症状,所以有人强调,如病人患有可能引起DIC的原发疾病,病程中出血症状又难以用其它原因解释时,应考虑到DIC的可能。
二、微血管栓塞引起脏器功能障碍
DIC病人尸检或活检时,常发现微血管(特别是毛细血管与微静脉)内有微血栓存在。它可由血小板组成,但典型的是纤维蛋白性微血栓。它们可以在局部形成,也可来自别处,从而阻塞微血管。在某些情况下,患者虽然有典型的DIC临床表现,但病理检查却未能发现阻塞性微血栓,这可能是由于体内凝血系统发动后纤溶系统同时被激活,使微血栓在生前或死后被溶解所致,也可能是纤维蛋白微血栓尚未完全形成,只有在电镜下才能见到。曾有人在研究猴的内毒素性休克与DIC关系时,获得这方面的证据。
微血管中形成的微血栓,可阻塞相应部位的微循环血流,严重时可造成实质脏器的局灶性坏死(图9-4)。严重或持续过久的坏死性病变可成为受累脏器功能衰竭的原因(图9-4)如果微血栓在肾脏形成,则病变可累及入球小动脉或肾小球毛细血管,严重时可出现双侧肾皮质坏死和急性肾功能衰竭,临床上表现为少尿、蛋白尿、血尿等。在肺部,可引起呼吸困难、肺出血,从而导致呼吸衰竭。消化系统的病变可导致恶心、呕吐、腹泻、消化道出血。肝脏受累时可出现黄疸及肝功能衰竭。内分泌腺的病变常见者为肾上腺皮质出血性坏死造成的急性肾上腺皮质功能衰竭,称华-佛氏综合征(Waterhorst-Friderichsen syndrome)。垂体坏死可导致席汉氏综合征(Sheehan's syndrome)。神经系统的病变可导致神志模糊,嗜睡、昏迷、惊厥等非特异症状,这些症状的出现并非是由一个孤立的局部病灶引起,而可能是由蛛网膜下腔出血以及微血管阻塞、脑皮质和脑干的多处出血所致。

图9-4 DIC的主要临床表现及其机制
DIC时由于凝血及纤溶的轻重程度不一,在不同的病人及病程的不同阶段可有不同的表现,此外,DIC范围大小不一所造成的后果也不同,轻者仅影响个别脏器的部分功能,重者可引起一个或多个脏器的功能衰竭即多器官功能衰竭,甚至造成死亡。
三、循环功能——休克
DIC,特别是急性DIC,常伴有休克。重度及晚期休克又可能促进DIC的形成,二者互为因果,形成恶性循环。
急性DIC常伴发休克,是由于毛细血管和微静脉中有广泛血小板聚集和/或纤维蛋白性微血栓形成,以致回心血量严重不足,再加上心肌损伤,广泛出血所引起的血容量减少等因素,使有效循环血量严重下降,心输出量减少,出现全身微循环障碍。与此同时,中心静脉压也往往降低,若肝和肺内有广泛微血栓阻塞,则又可相应地引起门静脉和肺动脉压升高。前者的临床表现为胃肠道瘀血、水肿,后者为右心排血障碍。此外,在DIC的形成过程中,由于凝血因子Ⅻ被激活,凝血酶增多和继发性纤溶的启动,可使循环血中Ⅻf、凝血酶和纤溶酶增多,它们均能激活补体和激肽系统,使激肽和某些补体成分(如C3a、C5a等)生成增多,激肽能使微动脉和毛细血管前括约肌舒张,从而使外周阻力显著降低;C3a、C5a等则可使肥大细胞和嗜碱性粒细胞脱颗粒,从而通过释放组胺而发挥与激肽类似的作用。这是急性DIC时动脉血压下降的重要原因。FDP的形成,加重了微血管扩张及通透性升高,这是因为FDP的某些部分(如裂解碎片A、B等)能增强组胺和激肽的作用,能使微血管舒张,因而更易产生休克。(图9-5)。
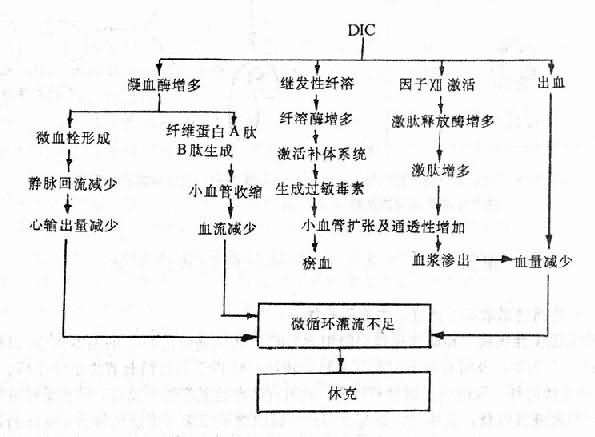
图9-5 DIC产生休克的机理
各种休克发展到一定阶段也往往可以伴发DIC(参阅《休克》)。
四、红细胞机械性损伤引起的微血管病性溶血性贫血
在DIC中有时可伴发一种特殊类型的贫血,即微血管病性溶血性贫血(microangiopathic hemolytic anemia)这种贫血除具备溶性贫血的一般特征外,外周血涂片中发现有某些形态特殊的变形的红细胞如裂体细胞(schistocyte),其外形呈盔甲形、星形、新月形等,统称其为红细胞碎片。这些碎片由于脆性高,故容易发生溶血。
目前认为产生红细胞碎片的原因虽然很多,但DIC是主要因素。其机制是当微血管中有纤维蛋白性微血栓形成时,在早期,纤维蛋白丝在微血管腔内形成细网;当循环中的红细胞流过由纤维蛋白丝构成的网孔时,常会粘着、滞留或挂在纤维蛋白丝上。这样由于血流的不断冲击,引起红细胞破裂(图9-6、图9-7)。在微血流通道发生障碍时,红细胞还可能通过肺组织等的微血管内皮细胞间的裂隙,被“挤压”到血管外组织中去。这种机械损伤同样也可使红细胞扭曲、变形和碎裂。这样就形成了上述各种畸形的红细胞碎片。所以在DIC病人中有时可以有溶血的一系列表现和实验室检查异常,外周血涂片中可出现较多的上述各种红细胞碎片。

图9-6 微血管病性溶血性贫血血片中能见到裂体细胞
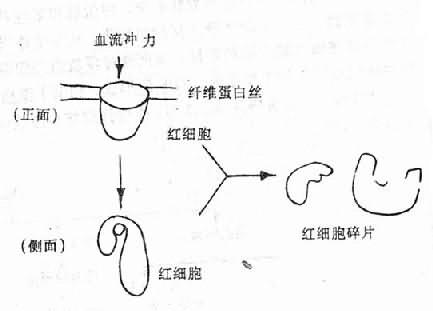
图9-7 红细胞碎片的形成机制
第五节 弥散性血管内凝血的防治原则
DIC的防治要采取综合措施,主要原则如下:
1.以严重感染引起的DIC为例,及时有效地控制原发感染病灶,对DIC的防治起着决定性作用。
2.
研究发现,肝素的抗凝效果与AT-Ⅲ相关。因为AT-Ⅲ是肝素辅助因子,肝素可与AT-Ⅲ的赖氨酸残基形成复合物,从而加速AT-Ⅲ对凝血酶的灭活,此外,AT-Ⅲ对血小板聚集也有一定抑制作用。DIC患者原先存在AT-Ⅲ减少或DIC本身引起的AT-Ⅲ减少均会影响肝素的抗凝效果。因此,有人认为,在应用肝素以前或同时,如能应用AT-Ⅲ制剂,则可提高肝素的抗凝效果。
3.重新建立凝血和纤溶间的动态平衡DIC时由于大量凝血因子及血小板消耗,因此在病情控制或使用肝素治疗后,以及在恢复期可酌情输入新鲜全血、冰冻血浆或纤维蛋白原等,以利凝血、纤溶间恢复新平衡。

 中医世家
中医世家 浦 标 网
浦 标 网 河南大学精品课程
河南大学精品课程 图书资料室
图书资料室